Public Channels
- # anonymous-question-box
- # ask-the-admin
- # day01-intro-genomics-biometric-model
- # day02-sem-study-design-phenomics
- # day03-quality-control-gwas
- # day04-heritability-and-gcta
- # day05-meta-analysis-prs
- # day06-genomicsem
- # day07-causal-modeling-mendelian-randomization
- # day08-sequencing-introduction-to-hail
- # day09-rare-saige
- # day10-pathway-and-gene-based-analyses
- # documentation
- # f-abcd-repronim-course-videos-instructions
- # f-creation-of-videos
- # general
- # python-intro
- # random
- # session-a
- # session-b
- # technical-support
Private Channels
Direct Messages
Group Direct Messages

@Jeff Lessem (he/him) has joined the channel

@Kumar Veerapen has joined the channel

@Tim Poterba (he/him) has joined the channel

@John Compitello has joined the channel

@Daniel Goldstein has joined the channel




@Test Student has joined the channel





@Sarah Brislin (she/her) has joined the channel


@Katie Bountress has joined the channel

@Peter Tanksley has joined the channel


@Charlotte Viktorsson has joined the channel


@Matthieu de Hemptinne has joined the channel


@Sam Freis (she/her) has joined the channel







@Stephanie Zellers (she/her/hers) has joined the channel



@Zoe Schmilovich has joined the channel

@Olivia Rennie has joined the channel

@Christina Sheerin has joined the channel

@William McAuliffe has joined the channel


@Francis Vergunst (he/him) has joined the channel




They web page with the video lectures for Sequencing and Introduction to Hail is at https://www.colorado.edu/ibg/international-workshop/2021-international-statistical-genetics-workshop/syllabus/day-8-wednesday
@Jeff Lessem (he/him) has renamed the channel from "sequencing-introduction-to-hail" to "day08-sequencing-introduction-to-hail"
@channel excited to see y’all tomorrow for our workshop session for sequencing and Hail. A few notes Remember to review the lecture on why sequencing analysis is important and why use Hail (https://hail.is/) :
https://www.youtube.com/watch?v=2N_VqmX22Xg&list=PL-A34BVyxWtXn9nxuj8Gk1yRfxhpdZ4y2
Review the python tutorial from Cotton. Why? Because Hail uses Python: https://www.youtube.com/watch?v=QIaunoHeP9Q
However, for the session, we will not be expecting too much coding from you. What we do want you to leave from the session is the ability to know what the pieces of code does? And obviously, to know how awesome Hail is for the analysis of sequencing data.
Finally, the practical sessions will be run on Google cloud services where we will provide a link and password to you tomorrow. All you need is a functioning web browser (and obviously internet access).
After the session, we will share the scripts and additional material (if any) via a github repo and a link on our Hail website.
If you have any questions in regards to the lectures that you have reviewed, in preparation for tomorrow’s session, and/or after tomorrow’s session, feel free to send us a slack message in this channel that’s solely for your teaching and learning experience in Sequencing and Hail.



I think a portion of the video differentiating genotyping from sequencing was cut out. Could you please clarify the distinction?
*Thread Reply:* Genotyping chips measure a small subset of the sites in the genome, which have been selected ahead of time to be sites of common variation that contain a lot of information. This is a cheap technology, but there’s a drawback - there’s not much information about rare variation.
The most common sequencing technology is high-throughput sequencing, also called short-read sequencing or shotgun sequencing. In this technology, the genome is chopped up into small (<150BP) chunks, each of which is sequenced separately. The billions of short reads are then aligned to a human reference genome, and after a processing pipeline (“variant calling”) you get information about every site in the genome (or exome) that contains the best-guess alleles for each individual, and some metadata about the sequencing process.
*Thread Reply:* Sequencing data is much bigger than genotype data, even for the same sample size, because so much more information is collected from each person. This makes it tougher to analyze.
*Thread Reply:* @Sage Hawn if you compare the slide deck available on the main page vs the video that was cut out, do you recall which slide was it?
*Thread Reply:* Thanks, @Tim Poterba (he/him) ! Always to the rescue!

*Thread Reply:* Re array vs. sequence data, I agree the main distinction by far is more rare variation in sequence. But sequence data also has much more info on non-SNP variants (indels). Do you agree @Tim Poterba (he/him)? And I’m not sure about the detection of larger CNVs and inversions/translocations in sequence data - can someone comment on that?
*Thread Reply:* That’s a great point! I’m often guilty of forgetting structural variation. Having worked on copy number variant calling from genotype data, it’s easy to find large deletions/duplications from genotype data, but sequencing data allows you to observe small deletions/duplications (at the level of a single base), and complex events like balanced translocations or rearrangements. But someone else should comment on why this is so important!
*Thread Reply:* For larger indels, you would have to use SV tools such as listed here : https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1720-5
I’ve had experience with pindel and breakdancer which the small nuances allow for things like transposable elements vs large indels.

*Thread Reply:* Thanks to you all - very helpful!

Hi! I might be completely missing the point here, but... How do PLINK (http://zzz.bwh.harvard.edu/plink/) and Hail relate to each other?
*Thread Reply:* They are both tools for managing and analysing genetic data. The difference is that PLINK is a standalone program that you run on the command line. Hail, in contrast, is a programming library that you use by writing Python code.
*Thread Reply:* Okay! And is there any recommendation of which tool is better? Or is that personal preference? For example, I believe the PGC (Psychiatric Genomics Consortium) mostly uses PLINK.
*Thread Reply:* PLINK is easier to learn and get started with, and works well with genotyping data on hundreds of thousands of samples and 10s of millions of variants.
*Thread Reply:* Hail requires more background to use, since you need to know Python, but it is ultimately more flexible since if it doesn't have the analysis you want to do, you can program it yourself (compared with PLINK, which can only do whatever commands it has built-in). Hail also scales better to sequencing data with 100s of millions of variants on millions of samples, since it can use cloud computing.
*Thread Reply:* The two tools have some overlap in functionality, but very different goals. PLINK is designed to be a set of prepackaged modules that work fast and well on genotype data (or well-QCed genotype calls that originally came from sequencing data).
PLINK only handles with the hard-call biallelic genotypes or genotype dosages. Sequencing data comes with more data (and more problems!) like allelic depth, genotype quality, and multiallelic variants.
Hail doesn’t have nearly as much pre-packaged functionality as PLINK does (you’ll see today, using Hail involves programming to ask the questions you want), but the expressiveness of Hail is required to handle sequencing data well, because every sequencing dataset has slightly different analysis needs.
As to which tool is “better” — that will depend on your application. My general rule of thumb is that if PLINK works for your project, use it! But if you are dealing with sequencing data or large datasets with genotype data (UKBB, for instance), Hail may offer a better experience.
*Thread Reply:* Thanks Mark, those are great answers!
*Thread Reply:* As an analogy, using PLINK is a bit like manipulating data using shell tools like awk and sort whereas Hail is more like manipulating data in R using dplyr.
*Thread Reply:* (Hail’s interfaces are heavily inspired by dplyr!)
*Thread Reply:* Thanks for the clarifications!
*Thread Reply:* I love this entire discussion! Tim will later be talking a little bit about this in our intro/primer to your practicals 🙂
Here are the instructions for connecting to the Hail workshop service! You can log in now, but please don’t start until you’re in breakout rooms so that everyone is going through materials together.


There have been a few questions about the meaning of the VCF fields (GT, AD, DP, etc.) Here is a description of those fields: https://gatk.broadinstitute.org/hc/en-us/articles/360035531692-VCF-Variant-Call-Format

See section 5, "Interpreting genotype and other sample-level information"
Many groups have questions about the SNP count exercises.
Question 1 - The reason why C/T and G/A SNPs occur at the same frequency is that they are the same SNP read from different directions! The ‘reference’ strand is arbitrary, so we should see each of these with the same frequency.
Question 2 - The various base mutations (C>T, T>A, etc) happen with different frequency due to the biochemistry of the nucleotides themselves. In particular, C>T mutations happen because when C nucleotides are methylated by nuclear machinery (called Cp), these bases look a lot like T nucleotides and replication errors from Cp > T happen higher frequency than any other substitution.

Quick descriptor of the ti/tv ratio: https://genome.sph.umich.edu/wiki/SNP_Call_Set_Properties
Also wikipedia: https://en.wikipedia.org/wiki/Transversion#Ratio_of_transitions_to_transversions

Apologies if I missed this during the session, how long will we have access to the notebook for to have a look at the practical again later?
*Thread Reply:* we’re going to answer that when rooms close. We’ll keep the rooms open for 2h or so, but if you want to run the practical after that, we can help you install Hail on your own computer, and you can download the materials!
@Isabella Loft If you could share the link to improving your knowledge?

*Thread Reply:* So I don't really have any good links, it is more about worked up knowledge by working in Python data stuctures, and a lot of boring parsing of files over the years.
But the MatrixTable remind me a bit of a nested dictionary visualised as a big table. Where it consists of a lot of nested dictionaries where the value for a key can also be a seperate table structure. The MatrixTable is visualised way better compared to raw data printed in python. Also dictionaries in python are not always the easiest to get you head around, and not always that efficient.
So with a quick google search
*Thread Reply:* Thank you for sharing!!! ❤
Breakout rooms and the workshop service will remain open for the next 2 hours!

The practical session was great! Very helpful, thank you 🙂

Hi, I might be missing this, but how do we download the files from the practical?
*Thread Reply:* The notebooks and data we used today are here: https://github.com/mkveerapen/2021_IBG_Hail/tree/main/resources
*Thread Reply:* Great, thank you!

*Thread Reply:* Would it be possible for you to upload pdf versions of the two notebooks with all the code printed so we can see the plots? An answer sheet to check all our answers would also be very helpful 🙂
*Thread Reply:* Here are PDF versions with all code run: https://github.com/mkveerapen/2021_IBG_Hail/tree/main/Materials/outputPDF
They don’t have the plots, though — the plots are interactive and work best as HTML. The GWAS tutorial on the Hail website (https://hail.is/docs/0.2/tutorials/01-genome-wide-association-study.html) has examples of similar workflows, though due to a bug the code output/plots are not showing up right now — if you check back in a few days it should be fixed.
*Thread Reply:* Thank you! Would it also be possible to upload an answer sheet with answers to all the questions in the two practicals?
*Thread Reply:* yes, that’s on my to-do list for today! 🙂
*Thread Reply:* Answer sheets are up: https://github.com/mkveerapen/2021_IBG_Hail/tree/main/resources
Hi all, we’re going to shut down the notebooks from session A in 10 minutes! The materials are available here: https://github.com/mkveerapen/2021_IBG_Hail/tree/main/resources
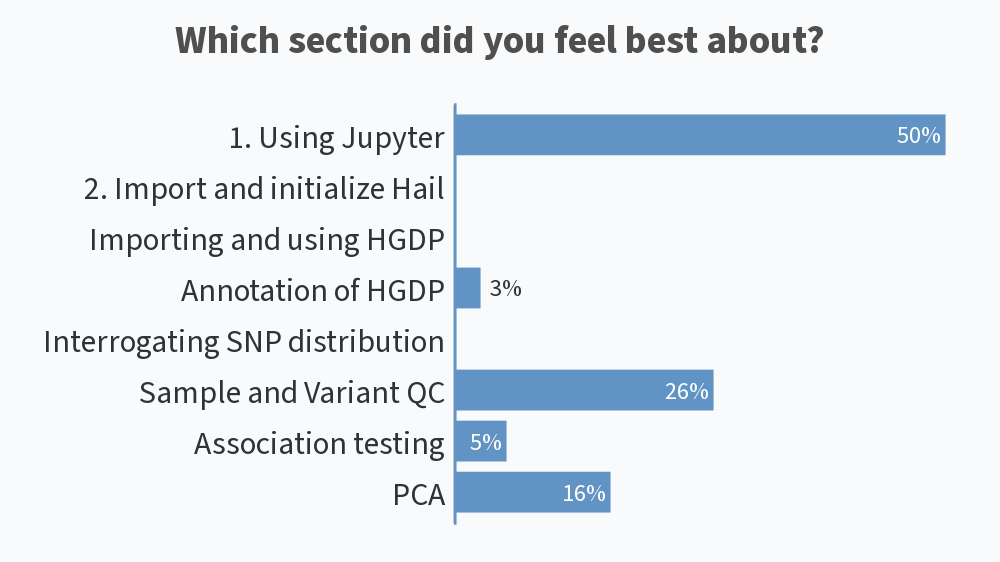
Results from this AM’s poll. Excited to see everyone for session B! 🙂
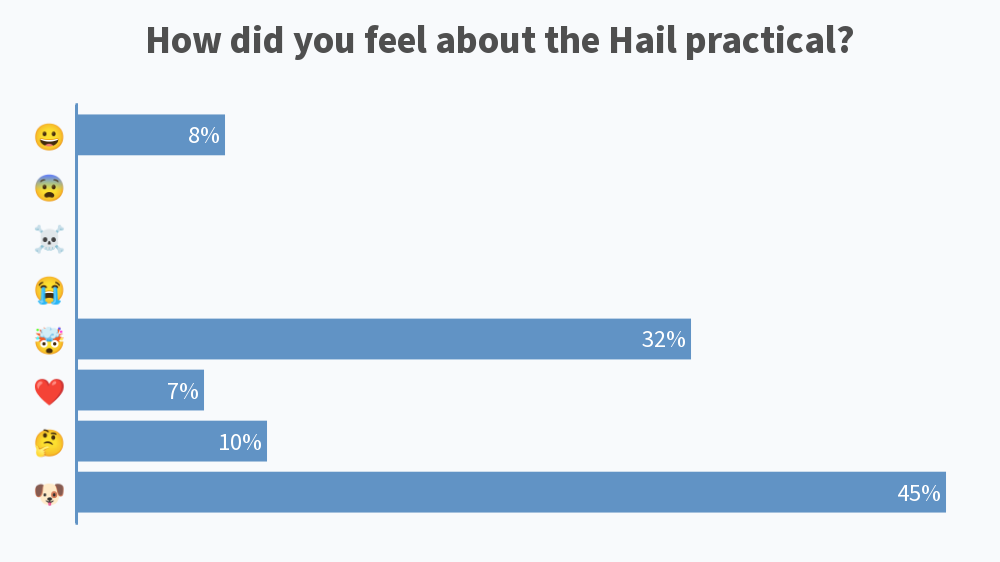
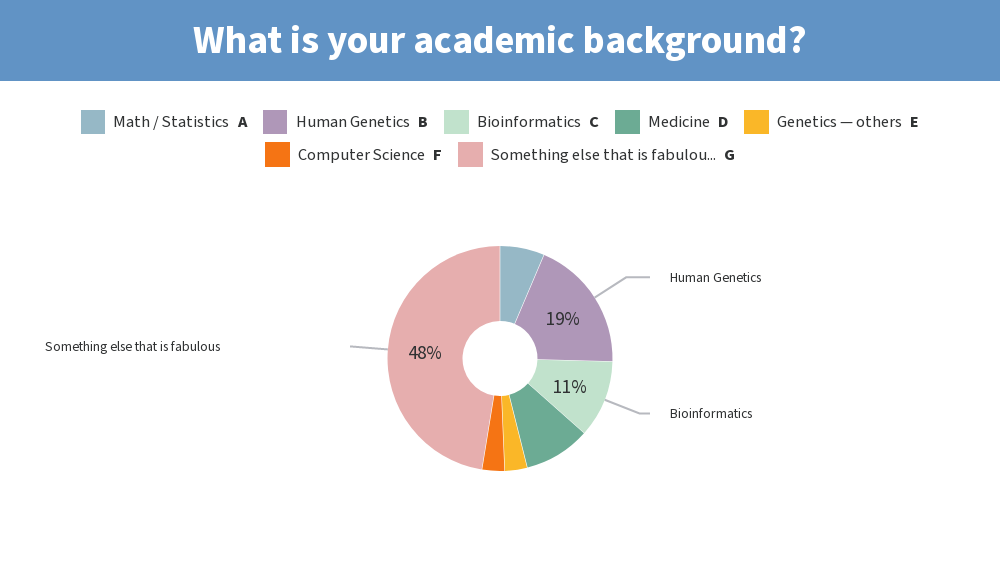
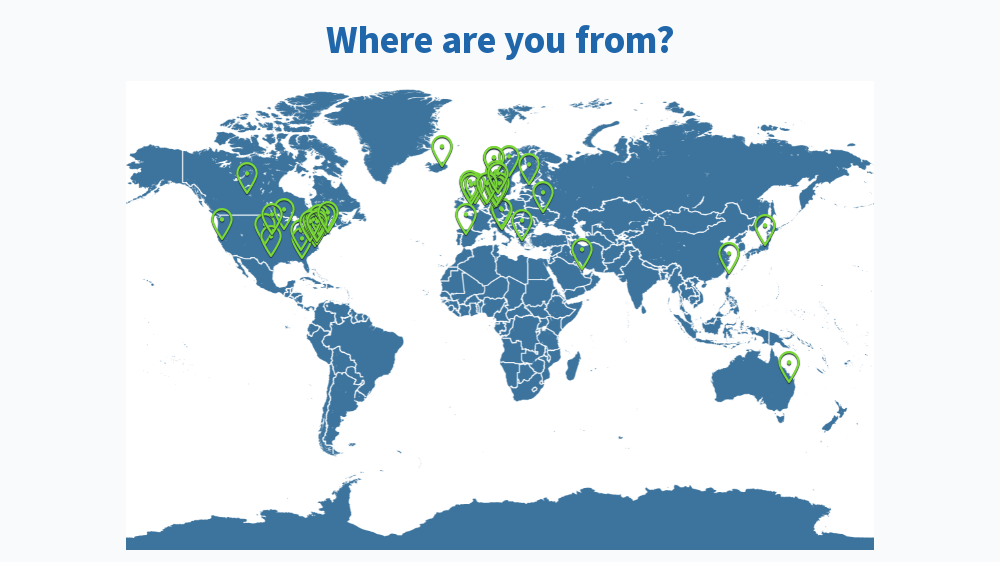
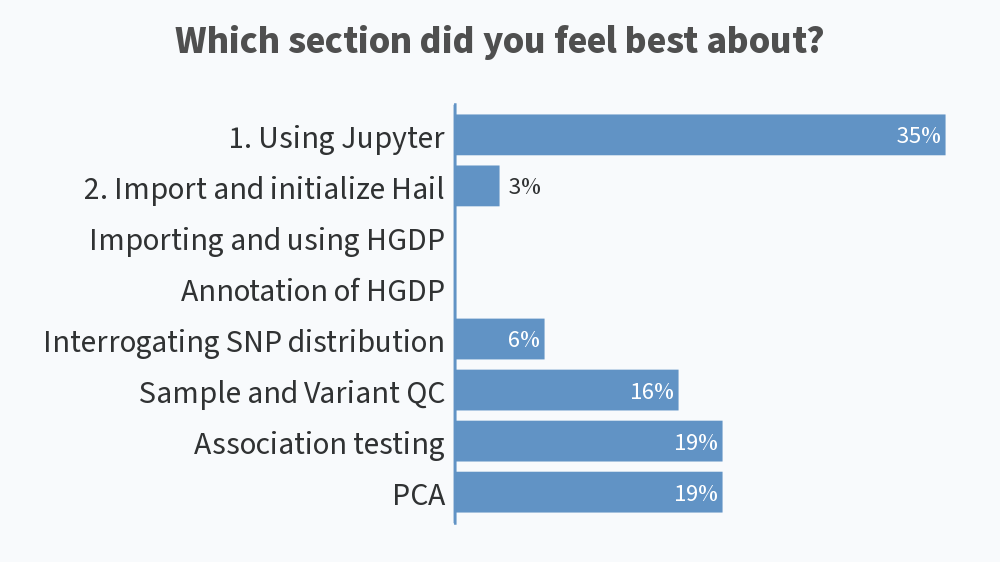
you should add [clinical] psychology to the 2nd poll for session B 😉

*Thread Reply:* I agree with Tim.
*Thread Reply:* maybe “social science” too
*Thread Reply:* Lol I just added “Psychology”
Instructions for connecting to the notebook server for today’s practical: https://boulder-workshop.slack.com/archives/C0201TSKBNE/p1623847680019900
 }
}
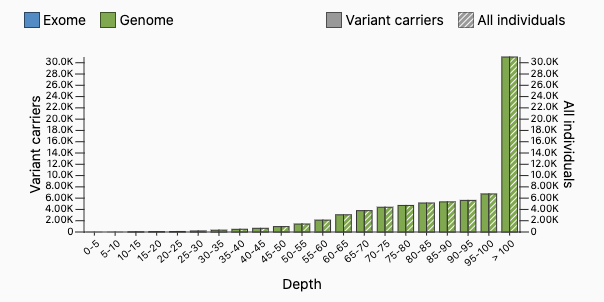
Some followup information about the HWE exercise — some questions have come up about how there could be sites with 100% of samples called heterozygous. What error mode could lead to this?
Here is one of these sites pulled up on the gnomAD browser: https://gnomad.broadinstitute.org/variant/1-125165544-G-T?dataset=gnomad_r3
We have theorized that a site could have 100% heterozygotes if there is a read mapping error upstream. Suppose that there are two 500-base-pair regions in different parts of the genome which are identical aside from a single base (one has a G, one has a T). When we use short-read sequencing, we’ll get some reads from both of these regions. If we align them to the same place in the reference genome (this is wrong), we’ll get a variant where every sample is a heterozygote. But there’s a way we can detect this — samples will have too many reads. The samples in gnomAD have mean depth 30, but the distribution for this site looks very different — most samples have more than 100 reads at this site!
Thank you for the Hail tutorial, it was very nice to use. Because our lab only has access to a slurm-based scheduler cluster, is it completely impossible to install hail on this type of computing cluster? It would be great to integrate hail into our workflows, but we will not have access to a spark cluster.
*Thread Reply:* I’m also wondering! Thank you!
*Thread Reply:* It’s possible but quite hard to run Spark (the way Hail runs on multiple nodes right now) on Slurm. However, it’s totally possible to run Hail on a single big node (32 cores or more) to do some heavy lifting. I think you could analyze 1000WGS in this model with no problem.
*Thread Reply:* okay perfect! We have access (on Compute Canada clusters) to nodes with up to 48 cores and 752G memory. Will definitely check with our team about working with it.
*Thread Reply:* that’s a boatload of memory 😱
*Thread Reply:* I think we would use a large proportion of our yearly allocation if we took the whole node there, can't say I've ever requested all of that!
*Thread Reply:* To be clear, 48 cores would be great and help your pipelines speed along, but you don’t need a huge amount of memory to run Hail. Something like 4-6G per core should be plenty.
*Thread Reply:* but it's not really possible to distribute these jobs across different nodes, right? Is that the limitation of slurm vs spark?
*Thread Reply:* with 48 cores would the invocation command be hail.init(local="local[48]") ?
*Thread Reply:* I think master="local[48]", though it’s possible your line would work as well. If you leave this argument out. The default is local[**] , which uses all available cores.
Notebooks and files for today’s practical are here: https://boulder-workshop.slack.com/archives/C0201TSKBNE/p1623861391032200?thread_ts=1623860342.031900&cid=C0201TSKBNE

thanks very much for the lectures and tutorials!
Hail is indeed a nifty platform to do many analyses, QC steps, and visualisations in one place. I am pretty impressed.
One minor note is that I might consider renaming the "impute" parameter in the hl.import_table function to something else, like "infer", because it sounds very much like imputing missing values (at least in the genetics context)!
*Thread Reply:* Thanks for this feedback! This is actually already on our list of things to change when we make a “breaking” version change (0.2 => 0.3 or 0.2 => 1.0). We can’t change it right now or it will break pipelines of existing users!
*Thread Reply:* sounds good. thanks!
We have updated the github repo containing all the materials used for teaching https://github.com/mkveerapen/2021_IBG_Hail We also included a link at the bottom if you’d like to download everything in a tar ball.
It was fantastic having such an engaging community to have an excellent teaching and learning experience with. Thank you everyone. We hope that you enjoyed yesterday and as much as we did! Please keep us posted with your adventures in Hail on either hail.zulipchat.com or discuss.hail.is.
Lastly, would also like to share the screenshots from Session B’s live polls in the following message
If you need the solutions to the notebook, Tim has posted it on the github repo 🙂