Public Channels
- # anonymous-question-box
- # ask-the-admin
- # day01-intro-genomics-biometric-model
- # day02-sem-study-design-phenomics
- # day03-quality-control-gwas
- # day04-heritability-and-gcta
- # day05-meta-analysis-prs
- # day06-genomicsem
- # day07-causal-modeling-mendelian-randomization
- # day08-sequencing-introduction-to-hail
- # day09-rare-saige
- # day10-pathway-and-gene-based-analyses
- # documentation
- # f-abcd-repronim-course-videos-instructions
- # f-creation-of-videos
- # general
- # python-intro
- # random
- # session-a
- # session-b
- # technical-support
Private Channels
Direct Messages
Group Direct Messages

@Jeff Lessem (he/him) has joined the channel
@Jeff Lessem (he/him) set the channel description: The place to ask anonymous (or signed) questions
@Jeff Lessem (he/him) set the channel topic: Anonymous and signed questions

To post an anonymous question, type /anon in the chat box. That will bring up a menu, and then click "Post Anonymously with AnonimityBot". That will bring up a dialog box. Choose the <#C01TLU7JAQK|question-box> channel, and enter your anonymous question.

*Thread Reply:* I can view this pinned message, Jeff!
*Thread Reply:* But I had to view the slack description/pin icon
@Jeff Lessem (he/him) has renamed the channel from "question-box" to "anonymous-question-box"

@matthew keller has joined the channel

@Lucía Colodro-Conde has joined the channel

@Tim Poterba (he/him) has joined the channel

@Katrina Grasby has joined the channel

@Kumar Veerapen has joined the channel





@Benjamin Neale has joined the channel


@Andrew Grotzinger has joined the channel

@Michael Neale has joined the channel

@Sarah Medland (she/her) has joined the channel


@Rebecca Richmond has joined the channel

@Adrian Campos has joined the channel



@Rob Kirkpatrick has joined the channel

@Baptiste Couvy-Duchesne has joined the channel

@Javier de la Fuente has joined the channel

@Abdel Abdellaoui has joined the channel

@Andrea Allegrini has joined the channel


@Emma Anderson has joined the channel

@Margherita Malanchini has joined the channel

@Daniel Goldstein has joined the channel


@Tetyana Zayats has joined the channel












@Jackson Thorp has joined the channel



@Christiaan de Leeuw has joined the channel


@John Compitello has joined the channel

@Alex Bloemendal (he/him) has joined the channel

@Elizabeth Prom-Wormley has joined the channel


*Thread Reply:* I'm not sure how anonymous it really is, but as the administrator, I cannot see who is using it. The company that publishes it claims they do not retain any data about who is using it. Worst case, some company in Israel will know which students are confused by H^2 and h^2.
*Thread Reply:* That was me trying this out!
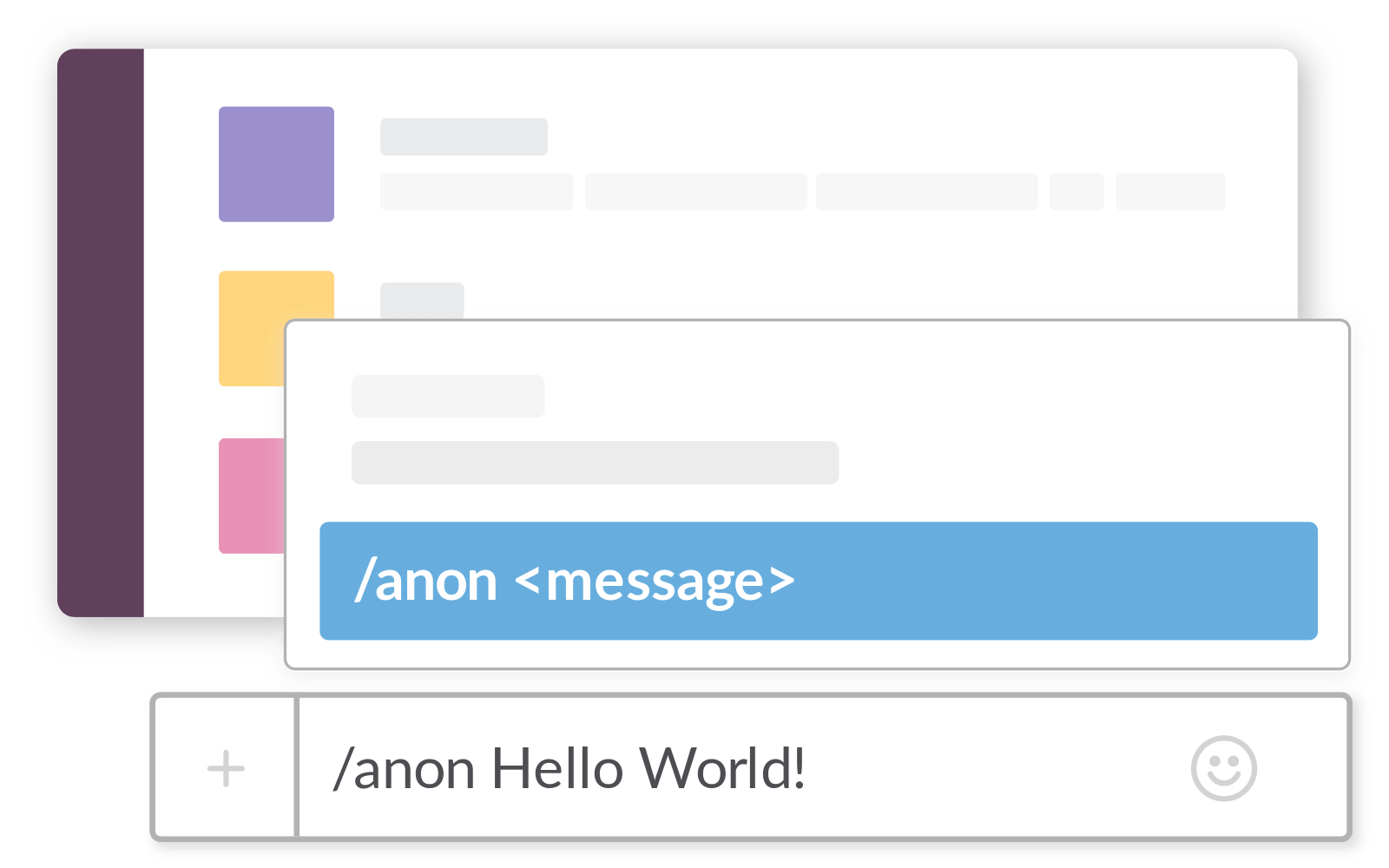
*Thread Reply:* You have to use the AnonymityBot by typing /anon. Otherwise your name is attached. Anonymous posting is limited to just this channel.
It was! I had no idea that we could do this! Pretty cool!
Do you see my pinned post? On mine it's scrolled away, which isn't really what "pinning" means to me.
*Thread Reply:* if you click the pushpin icon in the top, you'd find it
@Jeff Lessem (he/him) set the channel topic: Anonymous and signed questions, type /anon to post incognito!

@Test Student has joined the channel





@Sarah Brislin (she/her) has joined the channel


@Katie Bountress has joined the channel

@Peter Tanksley has joined the channel


@Charlotte Viktorsson has joined the channel


@Matthieu de Hemptinne has joined the channel


@Sam Freis (she/her) has joined the channel







@Stephanie Zellers (she/her/hers) has joined the channel



@Zoe Schmilovich has joined the channel

@Olivia Rennie has joined the channel

@Christina Sheerin has joined the channel

@William McAuliffe has joined the channel


@Francis Vergunst (he/him) has joined the channel


@Hilary Finucane has joined the channel
Hi, could the slides for Intro to Workshop and Basics of Stats Gen be made available? They're not currently posted on the syllabus Day 1.
*Thread Reply:* Yes - they should be in /faculty/ben/2021/NealeWorkshopIntro2021-upload.pptx
*Thread Reply:* I'll get them up on the website after the live session.
Adding onto the above would the slides for the next few days be available as well?
*Thread Reply:* It should be mostly available on the course page e.g. https://www.colorado.edu/ibg/international-workshop/2021-international-statistical-genetics-workshop/syllabus/day-8-wednesday
*Thread Reply:* > The slides used for this presentation are available as a PDF file.

I am running the R studio locally in my system and cannot open the dat files. Should I download any package in R?
*Thread Reply:* If you are running rstudio local to you, then you will have to download the files from the /faculty/conor to your computer and open them there.
There are lots of R packages that have been loaded. If you run ls /usr/local/lib/R/site-library/ on the server you will see the many that I have loaded.
For tomorrow, you will need OpenMX, umx, MASS, psych, and maybe some others.
That's why I generally recommend people use the hosted Rstudio, so they don't have to worry about that stuff during the practicals. Of course, you will need to worry about it for doing this stuff later on your own.

*Thread Reply:* If youre running R locally, please do not forget to install umx (R library) and OpenMx (R library). It is better to download the latter from the OpenMx site (something to do with optimization software used to estimate parameters). If you have any trouble downloading data from faculty/conor/Boulder2021, please send me an email requesting the material.
In question 1.4 of the practical by me (Conor) we read .... proportion is the additive genetic variance divided by the total genetic variance. That should be .... divided by the total phenotypic variance. The error has been fixed in the currect version of the pratical workbook (pdf and docx files in the folder: faculty\conor\Boulder2021. Apologies for the error (and thanks to Madhur Singh for pointing it out).

*Thread Reply: Thank you - with this in mind, could you just clarify where the division comes into it? I’m a bit confused as the answer only uses the multiplication calculation _(’the additive coded QTL explain 2.732% of the phenotypic variance (the variance component equals .02732*15.10257 = 0.412)._
*Thread Reply:* Ok: here we're predicting a single phenotype from a single QTL (measured genetic variant). In so doing, we want to know 2 things: 1) is the QTL associated (does it predict) and 2) if it predicts, how well does it predict. Associated with 1) is a null-hypothesis: b1=0, where b1 is the regression coefficient. Associated with 2) is the R-squared statistics, which is a proportion of explained variance to total variance.
*Thread Reply: Let's look at this in terms of question 1.4. of the practical. In the output you'll find the R^2 (R squared) which equals: Multiple R-squared: 0.02732, so 2.732 % of the phenotypic variance is explained by the QTL (T1QTL_A1 to be precise). The R^2 is explained variance / total variance. Now the R^2 is a proportion (and R^2 * 100 is a %). But we can work back from this by noting that the variance explained (NOT the proportion or the percentage), but the "raw" value equals R^2(total variance). So if R^2= y/x then y=R^2x ....
*Thread Reply:* Thanks for the question, Please see also the ppt presentation in which also I go into this stuff (the stuff being the concept of R^2 in linear regression).
*Thread Reply:* thanks very much, that’s very helpful
*Thread Reply:* Finally R^2 is a proportion of explained variance, Heritability ("narrow-sense" heritability) is the proportion of phenotypic variance explained by addititvely coded QTLs (snps). So narrow sense heritability is an R^2 statistic. BTW: for the concept of "additive coding" please see "backstory #1".
A followup question to the “more data or better data” discussion — how does the increasing number and availability of large national and hospital biobanks affect the way a group should allocate grant funding? On one hand, it seems to me that spending on analysis of these large (somewhat) public datasets would allow any individual group to do many cheap, well-powered studies. One the other hand, if everyone just uses the biobanks instead of generating new data, the scientific community is worse off because everyone is mining the same resources. When is it appropriate to spend on sample collection?
*Thread Reply:* Large centrally funded hospital(data) based biobanks Cheap well powered for sure, but in many cases (UKB and others) also opportunity samples that aren representative of the population. It may actually be time to think beyond opportunity samples and move to intergenerational samples (like ALSPAC) at the scale of UKB/Biobanks, preferralby linked to register data beyond healthcare. one funding mechanism I had been thinking about is every group can write a plan/study design for 250M$ we all then take a vote on what plan to execute and integrate good ideas from the losing proposal. data collection and study is run centrally data made available for all. Its trading 100 R01's/ERC’s for medium size biobank we all got a chance to design and a vote in…
*Thread Reply:* Thanks, Michel! The funding model you’ve proposed sounds like a great way to take advantage of centralized funding + data sharing, but also maintain community-driven experimental design.
in question 1.11, should the value obtained for dominance variance be the same as the value obtained for dominance variance calculated in 1.10? It is quite close but not the same. Why would this be the case?
*Thread Reply:* Good question. So we fit 3 model: 1) QTL additively coded QTLs 2) QTL dominant coded QTLs and 3) QTLs coded additively and QTLs coded dominantly. The decomposition should be 1) var(A); 2) var(D) and 3) var(A) + var(D). These variance components may differ (var(D) in 2 not equal precisely equal to var(D) in 3), but the differences should be small. This is because the correlation between the QTLA and the QTLD is close to zero, but not exactly zero. This is a consequence of my data simulation. It's a good sharp question, but the slight differences are not important here. If you want more details, let me know. Conor
Hi, is it possible to get a certificate of attendance after the workshop?
*Thread Reply:* I believe so. We’ll let you know soon
is there a reason why the videos to watch for today are over 4 hours instead of 2? some people may struggle to watch all of these before the session.
*Thread Reply:* I misunderstood my assignment and produced a ppt of 1 hour 45 minutes (it consistes of 4 parts, which can be viewed individually). My apologies for this. Hopefully you'll have time to view the ppts (later on). Conor
*Thread Reply:* So were we not supposed to watch it all for today? or were some for yesterday?
*Thread Reply:* I think you were supposed to watch 4 hours, but this won’t be the norm
*Thread Reply:* This was not doable for everyone I have spoken to about this and now people have to catch up today. Hopefully this will be taken into account in the future.
Apologies if this has been asked / answered already - but will the youtube videos remain online indefinitely?
Note that d≠0 implies dominance, where either d<0 or d>0 are possible.Note that d≠0 implies dominance, where either d<0 or d>0 are possible.
page 20 (backstory 2 Conor's practical) "Note that d=0 implies dominance, where either d<0 or d>0 are possible". Should be "Note that d≠0 implies dominance, where either d<0 or d>0 are possible" (thanks Joshua Pritikin) .
I hoped you managed to fit the ADE model using umx (my practical 7&8 june), The practical includes (ended with) the OpenMx script to fit the same ADE model. The OpenMx script is reproduced and annotated in Backstory #5 (in the practical workbook). So that may help to understand some of the details of the OpenMx script.
re. Day1 practical with umx, what does the Estimate of A_r1c1 (and others) from summary() mean? because it doesn’t look like VA? @c.v.dolan
*Thread Reply:* Hi the output is a bit cryptic because it is designed for multivariate analysis. The r1c1 bit refers to the row and column numbers of the matrix in which the estimate is located. HTH - Mike
*Thread Reply:* The Estimate of A_r1c1 (as you see it in the summary of the umx or OpenMx output) is based on the twin data is the additive genetic variance. In addition to this, we have the estimate of additive genetic variance based on the regression of the phenotype on the QTLs (coded 0,1,2). In the present simulated data (dataTw.dat), we find that the estimates are not really close in value. This is discussed on page 31 in the "answers to the questions" (p 31 in the practical "workbook"). conor
Hi! The two links of the GWAS slides (GWAS-part 1 & 2) in Day 3's webpage lead me to the QC's slides but not the slides for GWAS...any idea? Thanks!
*Thread Reply:* Fixed! thanks for letting me know.
could you please provide examples to further illustrate the difference between the common pathway model and the independent pathway model? how do we make decisions about which model to use? Thank you!@hermine.maes
*Thread Reply:* The 9 items in a psychological test are meant to measure (depend on) the phenotypic variable that I am interested in (say) "depression" or "depressiveness" - call it D. Note that D is a "latent" variable. I cannot observe it directly. So my ("psychometric") theory is the 9 items are phenotypes that depend directly and causally on D, where D is the latent variable of interest. If my D score is high, then I have a high probability of endorsing the item (say) "I feel blue almost everyday". My common pathway model theory works as follows. The items are directly and causally dependent on D, any genetic and environmental influence on D will result in genetic and environmental influence on the items (i.e., genes -> D -> item1, genes -> D -> item2, etc). In this model, the latent phenotype D mediates the genetic and environmental influence on the items. If I can demonstrate that this model fits well, then that is interesting because it supports the psychometric theory underlying my test (i.e., it support the idea that there is indeed a latent phenotypic variable D). A test of this is to compare the independent pathway model with the common pathway model. The latter is nested under the former. That means that we can use a likehood ratio test to determine whether the common pathway model fit adequately compared to the independent pathway model. Note that rejection of the common path model implies that there is no mediator D, and so casts doubt whether D actually exists.... For more on this please see Psychological Methods 2013, Vol. 18, No. 3, 406–43.
*Thread Reply:* see also Behavior Genetics volume 44, pages 591–604 (2014)
Just a comment -- so far I don't find the course accessible for someone new to BG methods. The level of assumed knowledge is too high, and even with experience using a variety of software packages the tutorials are too advanced to be meaningfully completed within the timeframe. The pre session intro videos are helpful but also highly technical for a newcomer and there is a missmatch between theory/conceptual background and application in the tutorials. Perhaps I misunderstood the purpose/ intended audience of the course...Anyway, just wanted to put that comment out there.

*Thread Reply:* I have to agree. Actually, I’m rewatching the videos for today and now things start making sense. Will start rewatching the ones for tomorrow, did it ahead and is impossible to remember the details I need for the practicals. I’m loving the course, do not take it the wrong way, virtual is harder, I appreciate all the effort from organizers and faculty involved.
Whoops, thought this was anonymous.... oh well
*Thread Reply:* Thanks for the feedback Francis! No worries about being critical (and outing yourself). We need to hear feedback. For someone completely new to BG, this will be a lot to take in. This workshop was also my first intro to BG back in 2004. It took some time and effort outside the course for it to start to sink in. You’re right that we’re trying to serve a fairly wide base. While we welcome people new to BG, this course isn’t designed solely as an intro to BG and BG methods; we’d like even seasoned veterans to be able to get something out of it. So hang in there.
*Thread Reply:* HI Francis: thanks for the feedback. I am glad I can address you by your name. I understand the problem. Many member of the faculty know what it is like to be overwhelmed in this course, i.e., many members followed the course as students in the past (including myself). I also know that many students follow this course twice. Like Matthew, I advise to to hang in there and get the exposure to the material. My ppt presentation was meant to be introductory. It seem long (1.45) but it actually consists of 4 short(ish) parts. I so not knwo whether you have had time to what it. It is supposed to be introductory (starting out with "simple" regression). Finally, if you manage to "hang in there", please feel free to pose your questions - any question!. E.g., if someone mentioned the term "parameter" and you're not 100% sure what that is (my experience once upon time), just ask! Thanks for the honest feedback!
*Thread Reply:* Thanks @matthew keller and @Conor for your comments and encouragement. The last two days have been easier as I've gotten more familiar with the software and the pre recorded videos weren't quite as long as the first couple of days which helped. Still a lot to take in but I think I'm going to make it! I wonder, though, if it wouldn't be helpful in future to start with the 'lighter' days without too much pre recorded/practical content, just to ease people in, and then go on to the longer days - like day 2 - which had 4h plus of pre recordings. Although I realise you may have other reasons for structuring the course the way you did. Anyway, just wanted to say I think you guys are doing a great job delivering this all online with so few hitches and keeping up with so many students.
Hi! Would it be possible to put all the relevant files and scripts for each day into one dropbox folder, and simply share a link to this folder? It would be very helpful to download the files/scripts on any computer anywhere. Thanks a lot!
*Thread Reply:* Hi - About a week or so the workshop all files are made available from the workshop website. During the workshop for most days all content is in a single folder and can be downloaded using an scp/ftp program.
*Thread Reply:* That being said we really want to encourage everyone to use the workshop R studio/sever to run the analyses as all required packages and programs have already been installed for you.
Please can you list the general format for what to type in the terminal and how to set the working directory in RStudio for each day? There seems to be slight variations each day (unless I am mistaken) which I am finding confusing p/anon
*Thread Reply:* Hi, You're right, there are many ways to do the same thing and can be confusing. We're trying to be more explicit with the instructions and now we're providing them in advance (see for example the Day 3 channel). Hopefully it helps!
Will our access to the workshop server remain after the workshop, or do we need to download anything we want to keep before the end of the workshop?
*Thread Reply:* Zip files containing everything from your home directory will be made available after the workshop, so you will be able to download all of you files.
Shortly after the workshop all files are made available from the workshop website. During the workshop files can be downloaded using an scp/ftp program. However, we really want to encourage everyone to use the workshop R studio/sever to run the practicals as all required packages and programs have already been installed for you.
*Thread Reply:* GWAS is a systematic evaluation of the evidence of association at approximately every variant; p-hacking is the phenomenon where re-analysis of the same dataset can bias tests to be more significant
*Thread Reply:* Perhaps most importantly, the GWAS standards of evidence are derived from the dimensionality of the association analysis [i.e., the effective number of tests]. See Pe'er et al https://pubmed.ncbi.nlm.nih.gov/18348202/ and Dudbridge and Gusnanto https://pubmed.ncbi.nlm.nih.gov/18300295/ for estimation of these which was followed up with Li et al. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3325408/
For a p-hacked GWAS, we could imagine looking at a single SNP, testing it, comparing it to 0.05 and then publishing when we find one that hits that threshold.
Even the multiple testing corrections don't necessarily manage the issues of shifting which covariates to include, subset analyses or other ways of trying to squeeze out more association evidence that could be considered as p-hacking
*Thread Reply:* I would even say that GWAS was an antidote to the widespread p-hacking that many of us suspect was happening in the pre-GWAS candidate-gene literature. GWAS's standards of genome-wide significance and independent replication was something that human genetics sorely needed in the mid- to late-2000s.
This might be a naive question,..but I wonder what happens to the genetic material (e.g., saliva sample) after genotyping is completed? are they stored somewhere or just be thrown away? what is the common procedure?
*Thread Reply:* Not at all naive. So, I think there are two things to address. First, after DNA isolation takes place, the "left over" saliva within the assay is discarded as biohazard waste. Second, there is generally always large concentrations of DNA that are left over after genotyping (this can be a good thing). Generally, investigators anticipate this and will store DNA in freezers for long term storage and use in the future as opportunities arise. It is advisable for an investigator to anticipate that they will have additional DNA and put discussion of this point in their informed consent documentation and be VERY CLEAR with participants about what they intend to do with the DNA afterwards (e.g., storage, for how long, who will DNA/genotype data be shared with in the future). Generally, in the US (the country I am most familiar with) Institutional Review Boards (IRB) have a pretty rigorous review of consent documentation with respect to DNA collection, storage, and use. They will guide investigators to best practices at that time. I feel comfortable saying it is equally (if not more) rigorous with IRB in Europe and Australia. As with many genetics-related things, this conversation is evolving. One reference (let me know if you have trouble accessing) https://pubmed.ncbi.nlm.nih.gov/30864046/

I am not a statistician by training, but have been trying to improve my understanding of what's going on "under the hood" with various statistical methods. I usually seem to hit a barrier with the matrix algebra part. I understand the basics like matrix multiplication, but where I get stuck is: 1. Why does linear dependency between rows of a matrix affect whether or not it can be inverted? 2. Why does inverting matrices seem to be a major/important part of what so many statistical methods do? Can anyone help explain this part or suggest a good beginner-friendly resource for understanding it?
*Thread Reply:* Suppose someone goes to the supermarket and buys a pound of apples and a pound of cherries, and tells you that they cost $8. From this data, you don’t know whether they each cost $4, one costs $5 and the other $3 etc.
Luckily, another person comes out with a pound of apples and half a pound of cherries, which cost them $5. Now we have the information to figure out that apples are $6/lb and cherries $2. While a little algebra with simultaneous equations would do it, we can do the same thing in matrix form, which I’ll describe in a bit.
Suppose instead that we had been unlucky, and the second person had come out with 2 pounds of apples and 2 pounds of cherries and the cost was $16. See how this doesn’t help us to unambiguously figure out the prices of each fruit? This is because here the second person’s purchases were a linear combination (2x) the first person’s.
In matrix form (which will be a bit crappy in this post, sorry:
1 1 apples = 8 2 2 cherries = 16
Consider the left hand side as a 2x2 matrix and a 2 x 1 vector, and the right hand side as a 2x1 vector. Here the weight matrix (the first one with 1s and 2s) is singular, it has a determinant of 1x2 - 1x2 = 0. No inverse due to linear dependency.
1 1 apples = 8 1 .5 cherries = 6
Here we are in business, as the inverse exists. Use R to check that out, find its inverse and then pre-multiply the right hand side by this inverse. You should get the same answer as above.
*Thread Reply:* Great question! And to make the link to statistics, the most fundamental example is linear regression, in particular linear least squares (also historically the first, dating back to Legendre and Gauss around 1800). Here you are trying to fit a linear function to data. You can express the problem as a linear system as in Michael’s example, but it no longer generally has an exact solution: there is generally no linear function that perfectly fits the data. So instead of just solving the system, you minimize the sum of squared residuals. The mathematical expression of this “least squares solution” involves a matrix inverse; the so-called normal equations generalize exact solution of a linear system (and can indeed be viewed as a direct generalization of the matrix inverse, the so-called https://en.wikipedia.org/wiki/Moore%E2%80%93Penrose_inverse|pseudoinverse).
Other examples of matrix inverses in probability and statistics include conditionals of the multivariate normal distribution, partial correlation, and many more. Multivariate statistics is built on linear algebra, and inverting a square matrix is fundamental to linear algebra just as division is to arithmetic.
My favorite introduction to linear algebra is Gilbert Strang’s course on MIT Open Courseware and
*Thread Reply:* Just one final reply to this question. In simple regression, the slope b1 = cov(x,y)/var(x) where x and y are vectors. It could also be written as b1 = SSxy/SSx where SSxy is sum of squared cross-products and SSx = sum of squared products (the latter equation just cancels out the “/n-1” term that exists in both the numerator and denominator). In multiple regression, X is now an n-by-p matrix (think of X being made up of p column vectors of length n for each 1…p predictor), and the corresponding formula for a vector of b’s is ((X’X)^-1)(X’Y). The left part ((X’X)^-1) is the matrix equivalent of dividing by SSx in the simple regression formula. Therefore, (X’X), which is a square matrix of dimension p-by-p, must be invertible for this to work (because ^-1 denotes inversion).
*Thread Reply:* Good point — those are the normal equations I alluded to, and I agree they only work in the “regular case” when X’X is invertible. That fails if the design matrix X has rank < p, which happens anytime n < p. In that case you can try to “regularize” the problem, which begins to connect to statistical learning. The simplest way to regularize is to add a small multiple of the identity matrix to X’X before inverting it, which is called ridge regression. Regularization is great for prediction but tends to shrink coefficients toward zero, which is challenging for inference and interpretation.
What happens (and what does it mean) when HWE is violated? do we excluded the SNPs that violate HWE from analysis?
*Thread Reply:* It most often reflects a technical problem with the clustering of the intensity data or sample contamination. Typically variants when the HWE Pvalue <=1e-6 are discarded. Because there is so much LD (auto-correlation) among nearby SNPs any real signal that would have been seen at a discarded SNP should be seen in SNPs in high LD in the surrounding region so dropping these variants is not as bad as it sounds. These variants can also be imputed back in later. Another option if there were lots of SNPs being dropped is to re-cluster the raw data or run some in-house genotyping to check for contamination.
*Thread Reply:* As Sarah notes, large observed deviations from HWE almost always reflect a technical or measurement issue, which serves as a very useful QC criterion. Subtler true deviations, however, arise anytime a population is not mating randomly (panmictic).
Population structure generally leads to decreased heterozygosity and excess homozygosity in the total population relative to HWE, which is one way to think about F_ST, as discussed in my lecture (Wahlund effect). Consanguineous mating or inbreeding also leads to decreased heterozygosity. In both cases, a pair of uniting gametes are more likely to carry the same allele than a pair of random gametes are.
A rarer scenario is that of excess heterozygosity, which occurs among first-generation admixed individuals from previously diverged groups. Here uniting gametes are more likely to carry different alleles than a pair of random gametes are.
Finally, even a single generation of random mating restores HWE! https://en.wikipedia.org/wiki/Hardy%E2%80%93Weinberg_principle|Wikipedia is quite good here.

*Thread Reply:* As a consequence, it’s possible to be too strict in filtering out SNPs that violate HWE, especially when doing PCA or other ancestry inference. If the deviation is real and not just a technical artifact, then the SNP is ancestry-informative, and thus key to the inference. That’s why the HW p-value threshold for QC is typically quite extreme.
*Thread Reply:* Another source of true HWE deviation is from ascertainment or selection bias. Research cohorts tend to be better educated and healthier than the population and thus they will be slightly enriched for alleles that increase health or intelligence. A more extreme instance of this occurs if disease cases and controls are collected and genotyped separately, as then there will be HWE deviations in any variant that separates cases from controls.
Is it possible to run GWAS with ordinal or categorical phenotypes? Perhaps in software other than PLINK?
*Thread Reply:* you can in gwsem (https://cran.r-project.org/web/packages/gwsem/index.html) which @Brad Verhulst will be talking about!

*Thread Reply:* There is this resource here: https://www.sciencedirect.com/science/article/abs/pii/S0002929721001038 which is implemented in SAIGE (taught on Day 9)

I know it has already been said that transcripts will be made available after the workshop. For the videos with un-edited captions, would it be possible for someone to edit the captions? They can be quite confusing when inaccurate (and also distracting while watching the videos). This would be incredibly helpful. Thanks!
This is a very basic question and you probably have mentioned it in the lectures and I missed it, but how do I figure out which build are the SNP data mapped to, by looking at the map file?
*Thread Reply:* Hi - you can't tell from the map file alone. If you search a couple of the variants against build 37 in the genome browser and the possitions match then the positions are from build 37. If they don't match then check against build 36 and build 38 till you find a match
*Thread Reply:* Chromosome numbers prefixed with "chr" ("chr1", "chr2", etc) instead of just numbers ("1", "2") is sometimes a useful clue that the SNP data are mapped to build 38.
*Thread Reply:* Agreed on the CHR3:43790 vs 3:43790 naming format - some old files are still on build 36 though so it's worth a check
Hi! We discussed on today’s session this for a while, but I am still left with some questions so I’ll just post them here. Basically, I am wondering what is the best strategy to match cases and controls, especially in a hypothetical situation where we have way more controls than cases? 1) What is the best proportion of cases vs controls? Is it 1:4? 2) What should we have in mind when choosing a subset of controls to match the cases? Also, in a situation where we may have more cases than controls, what would be the best strategy to match them? Thanks a lot!
*Thread Reply:* I wasn’t at today’s session, but one way to get at (1) is to consider the effective sample size, which governs power in a case-control association study. (This operates via the non-centrality parameter; Matti Pirinen’s notes are nice, and the appendix of this paper gives a full treatment.)
With N0 controls, N1 cases, N = N0 + N1 total samples, and phi = N_1 / N the fraction of cases, the effective sample size is
N_eff = N phi (1 - phi) = 1 / (1/N_0 + 1/N_1).
[Exercise: show these two forms are the same.]
Now to answer your question, it depends on your constraints! If N is fixed (say if your genotyping budget is the limiting factor), the optimal phi = 1/2, which makes Neff = N/4; in other words, a 1:1 case:control ratio. On the other hand, if you have a fixed number of cases and unlimited controls, the more the merrier! Here the second form is useful and shows Neff approaches a limit of N1 as N0 becomes large.
*Thread Reply:* Your second question can be answered at several levels. You first want to match at the technical level: ideally your cases and controls would be genotyped or sequenced on the same platform, e.g. using the same array/chip. If not, batch effects (especially from imputation, it turns out) may create spurious signals and even dominate your analysis — although a pipeline combining stringent QC and careful imputation can mitigate these, as we showed in this preprint.
You also want to match at the level of ancestry, to avoid the problem of population stratification as discussed in my lecture. That does not mean matching one-for-one; as long as the ancestry distributions are reasonably well-matched, inclusion of PC covariates should work reasonably well in most settings.
In fact, both pieces can be assessed to some degree using PCA, as genome-wide batch effects will also tend to manifest as driving a component or two. Look at multiple bi-plots (beyond just PC1 vs PC2), plot cases and controls in different colors, and look for overlapping distributions without systematic deviations. (If they mostly overlap, you can prune outliers.)
Finally, depending on your trait, you may want to consider matching on age, sex, and perhaps other environmental factors. Here again, inclusion of covariates allows you to avoid having to match one-for-one.
*Thread Reply:* It’s important to realize that is you go beyond what Alex is suggesting here and you decide to match controls on heritable traits (and really many things are heritable) you influence and alter the outcome of future analyses on the relationship between traits that rely on the GWAS you are doing. The same holds for using convenience controls like “blood-donors” or “people with a different disorder in the same hospital” those selections of people have an allele frequency other than the whole population, and so while they could serve as a control when you want to find top hits for your traits, their selection as controls does influence downstream analysis!
*Thread Reply:* Some mortician support for my argument: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3781001/
*Thread Reply:* As an addendum- practically speaking, not much increase in power after your controls reaches 4 to 5 times your number of cases.
I'm wondering how to use regression estimation methods other than linear and logistic (e.g. Poisson, zero inflated) to conduct GWAS. In particular, when the assumption of normality of the phenotype is not meant. Is that an option in Plink?
*Thread Reply:* You can deal with ordered data in gwsem (https://cran.r-project.org/package=gwsem) there are methods for censored time to event data as well (just one specific one I read: https://www.medrxiv.org/content/10.1101/2020.09.04.20188441v2.full) other faculty like @joshua pritikin or @Brad Verhulst van tell you more about gwsem when the wake up.
I understand that using genetically related individuals could bias estimates of heritability using GREML, but at the same time having genetically related people I would assume would also be helpful. Do researchers ever use adopted twins to obtain heritability using GREML? I feel like this would help with the bias of shared environmental factors that can't be measured, while still improving the confidence intervals around heritability? In other words, would there be a benefit of using adopted twins when using GREML, or am I missing something?
*Thread Reply:* By adopted twins, I suppose you mean twins reared apart? If so, thankfully (for the twins) but unfortunately (for science), this just doesn’t occur much anymore, and even when it happened back in the day, there were very few of them, so sample sizes would be far too small to do GREML analyses on them. That said, there are approaches for using close relatives in GREML - see the Zaitlin paper from 2013 I think in PLoS Genetics
*Thread Reply:* Here's a link to the Zaitlen et al. paper: https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1003520 . Also, I myself would not say that including close relatives in a GREML analysis "biases" the estimate of h2SNP. Instead, it changes the interpretation of h2SNP. When there are no close relatives in the sample, h2_SNP is interpretable as the proportion of phenotypic variance attributable to the common SNPs on the genotyping array, and to markers that are in LD with those SNPs in the population. But that interpretation comes apart when you have close relatives in the sample, because genotypic similarity between relatives w/r/t common SNPs also potentially tags resemblance w/r/t rare linked mutations within the pedigree.
*Thread Reply:* Note that Zaitlen et al. (if memory serves) explicitly estimate a shared-environmental variance component. They also estimate two genetic variance components: one is variance attributable to the SNPs on the array (and markers in LD with them), whereas the other is a pedigree-type variance component based on the expected genetic resemblance among people (which is zero for people who aren't in the same family).
can you clarify what the working directory for rstudio should be each day?
*Thread Reply:* The working directory that you will be doing your practicals should be a folder within your own main main folder. Each speaker provides instructions in their day-specific channel for where to find files. You should be copying the files from a faculty folder to your folder. Once all the files are copied from a faculty account, then you can set up your working directory in R to point to the place in your own account. If it helps, just reach out to me directly. I'm happy to set up a time/zoom to ensure you're all set up prior to a lecture.
Another naive question but pls can someone say what is the difference between haplotypes and LD? thk you
*Thread Reply:* In basic terms, LD (linkage disequilibrium) refers the occurence of specific combinations of alleles at two or more linked loci more frequently than expected by chance. Linkage disequilibrium is the result of recombination during meiosis. Consequently, certain groups of alleles are more likely to be inherited together as blocks of alleles, or haplotypes. Haplotypes are a group of alleles at closely linked loci. It is because of LD and haplotypes that we are able to engage in imputation. A couple of references that may be helpful https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5124487/pdf/nihms-831771.pdf
*Thread Reply:* https://www.nature.com/articles/nature04226
*Thread Reply:* Another way to think of it is that haplotypes (a segment of a chromosome passed down from an ancestor) is the fundamental CAUSE of LD. If there is a particular haplotype that is long and shared by many people in the population, the alleles that exist on that haplotype co-occur at rates above chance, hence there is LD between the SNPs in the region defined by that haplotype.
How do we define fixed and random effects in the context of genetic analyses? I have seen there are many many definitions of them that are mostly field-dependent on the internet
*Thread Reply:* Great question. This is indeed not straightfoward and the answer do depend on your study design, on whether you believe there is a true value somewhere (e.g. the effect of a SNP in a given population) that you really want to estimate, or also on whether you have enough data or not. Modelling the effects of millions of SNPs as random effects can be convenient when you don't have enough data (sample size) to directly estimate them. It's important to keep in mind that the distinction between fixed and random is mostly for modelling purposes and that effects are not intrinsically "fixed" or "random". I can elaborate more if you have a more detailed example to share. Thank you!
*Thread Reply:* Here is a take on random vs fixed that relates specifically to GCTA (and also to rare variant testing).
*Thread Reply:* I don't blame OP for being confused about fixed versus random effects, in the context of genetics or otherwise. The terminology is just plain messed up: https://statmodeling.stat.columbia.edu/2005/01/25/why_i_dont_use/ . I don't think I really grokked the distinction until I was in my last year as a PhD student.
*Thread Reply:* Suppose I have 10 QTLs (additively coded) and a phenotype measured in N=10000 people. I can carry out a straightforward regression analysis: phenotype = b0 + b1QTL1+b2QTL2+ ...+ b10QTL10 + e. In this model I obtain estimates of b1 to b10, which I can test (e.g., H-null b1=0). This is standard fixed - effects regression where b1 to b10 are fixed effects. I can also test the omnibus null hypothesis b1=b2=...=b10=0. Ok now suppose that we have 1000000 QTLs (yes, that is a million). I cannot do the fixed effect regression because there are more predictors than persons (one million vs 10000). This is where the random effect model comes in. Rather than estimate the individual b1 to b1000000 as fixed effects, I introduce a distributions for the b's. E.g., a common assumption is b~N(0,sd(b)): the regression coefficients are distributed normally with mean zero and standard deviation sd(b). So rather than estimating individual b parameters I am estimating the variance. Note that the omnibus null hypothesis is sd(b) = 0, which makes sense: if all the b parameter are zero, the variance (or standard deviation) is zero. I think that this is the basis of GCTA and the SCAT test (in rare variant research, which returns later on in the course).
*Thread Reply:* If you want a R based demo, let me know.
*Thread Reply:* BTW: I think it's SKAT (not SCAT) test. sorry about that!
*Thread Reply:* In the above b~N(0,sd(b)) mean the b's are distributed normally with mean 0 and standard deviation sd(b).
*Thread Reply:* Ok sorry for the length of this. In the above b~N(0,sd(b)) means the b's are distributed normally with mean 0 and standard deviation sd(b).
*Thread Reply:* But now you may think: why mean zero? (in N(0,sd(b)). That is because I assume that the average effect (b) is zero. That is averaged of the 1000000 b's. Do we have to assume this? No, there is also a version of this in which the mean is estimated ("burden test"). There's alot more on this in rare variant part of this course.
*Thread Reply:* Question: Doesn’t the GREML approach have it backwards? Shouldn’t we be treating SNP effects as fixed (they don’t differ across samples!) and the GRM as random rather than vice-versa? In practice, maybe it doesn’t matter, but logically it seems backwards. @Loic Yengo
*Thread Reply:* I suppose that the status of the predictors (random variables vs fixed variable) is a consideration, but I interpreted the question as fixed vs random w.r.t. the regression parameters. My answer was based on my understanding (eh....) of SKAT vs burden (and combined Skat-burden), Skat and GCTA are closely related. As long as my answer to the student is not BS, I am happy.
*Thread Reply:* GREML do assume SNP effects (beta) to be random and only concentrates on estimating their variance. I agree with you that in reality sampling of genotypes is the randomness generating process at play here.
*Thread Reply:* Thank you so much for this! I took a while to figure out all the details in your replies, but I think I am kinda up to pace on what they are in genetic studies. I have to agree with Rob that it's difficult for a beginner, while searching for the answer I met that post from Gelman that probably confused me even more. My question was originally raised from LMMs and GCTA, but Conor's explanation with GCTA was really really useful, thank you! I don't know if the offer is still valid, but a mini-simulation in R would be really helpful (if it doesn't take too much time on your side).
*Thread Reply:* library(nlme) # use lme to do the random effects regression control=lmeControl(msMaxIter = 2000) # lme parameter # set.seed(111222) NQTL=50 NQTL1=NQTL+1 pA=runif(NQTL, min=.05, max=.5) meanb=0 # mean of regr coeffs sdb=1 # stdev of regr coeffs b=rnorm(NQTL, meanb, sdb) # regression coeffs N=2000 # sample size # ToyDat=matrix(1,N,NQTL1+1)
simulate the QTL... uncorrelated in linkage equilibrium. Simple toy exmaple.
for (i in 1:NQTL) { p=pA[i] q=1-p gf = c(p^2, 2pq, q^2) # genotype freq assuming Hardy Weinberg Equil. ToyDat[,i]=sample(c(0,1,2),N, replace=T, prob=gf) }
create the phenotype ... part 1 ... due to the QTLs
for (i in 1:N) { predicted=0 for (j in 1:NQTL) { predicted=predicted + b[j]**ToyDat[i,j] } ToyDat[i,NQTL1]=predicted } sdpred=sd(ToyDat[,NQTL1]) # sd due to the QTLs #
add 50% residual variance, normally distributed "environmental" variance
environmental = rnorm(N, 0, sdpred) # using sdpred implies 50% (QTLs) vs 50% (env) ToyDat[,NQTL1] = ToyDat[,NQTL1] + environmental # colnames(ToyDat) = c( paste('Q', 1:NQTL, sep=""), # produces Q1, Q2, ... etc. 'pheno','unit') # variable names. ToyDat=as.data.frame(ToyDat)
ok data ready to go: fixed effects model ... this is just standard regression
f_model=lm( pheno~Q1+Q2+Q3+Q4+Q5+Q6+Q7+Q8+Q9+Q10+ Q11+Q12+Q13+Q14+Q15+Q16+Q17+Q18+Q19+Q20+ Q21+Q22+Q23+Q24+Q25+Q26+Q27+Q28+Q29+Q30+ Q31+Q32+Q33+Q34+Q35+Q36+Q37+Q38+Q39+Q40+ Q41+Q42+Q43+Q44+Q45+Q46+Q47+Q48+Q49+Q50, # there must be an easier way! data=ToyDat) #
the explained variance should be about 50% (R-squared)
# estb=fmodel$coefficients[2:NQTL1] mean(estb) # about zero as meanb =0 sd(estb) # about equal to one as sdb = 1 plot(b,estb,type='p', xlab='true b', ylab=' est b fixed') summary(fmodel)$r.squared # should be about 50% # rmodel=lme(pheno~1, random=list(unit=( pdIdent(~Q1+Q2+Q3+Q4+Q5+Q6+Q7+Q8+Q9+Q10 + Q11+Q12+Q13+Q14+Q15+Q16+Q17+Q18+Q19+Q20 + Q21+Q22+Q23+Q24+Q25+Q26+Q27+Q28+Q29+Q30 + Q31+Q32+Q33+Q34+Q35+Q36+Q37+Q38+Q39+Q40 + Q41+Q42+Q43+Q44+Q45+Q46+Q47+Q48+Q49+Q50-1))),control=control,data=ToyDat,method='ML') summary(rmodel) # rmodel$coefficients$random$'unit'->estb2 plot(estb,estb2,type='p', xlab='est fixed b', ylab=' est random b')
based on all QTLs
# PRS1=rep(0,N) for (i in 1:N) { for (j in 1:NQTL) { PRS1[i]=PRS1[i]+b[j]**ToyDat[i,j] }}
based on 10 QTLs
PRS2=rep(0,N) for (i in 1:N) { for (j in 1:10) { PRS2[i]=PRS2[i]+b[j]**ToyDat[i,j] }} # ToyDat_ = cbind(ToyDat, PRS1,PRS2) #
What if we add the PRS1, contains all the genetic info. So sdb estimated should be zero
PRS1 is the fixed regressors the QTL are the random regressions.
# rfmodel1=lme(pheno~PRS1, random=list(unit=( pdIdent(~Q1+Q2+Q3+Q4+Q5+Q6+Q7+Q8+Q9+Q10 + Q11+Q12+Q13+Q14+Q15+Q16+Q17+Q18+Q19+Q20 + Q21+Q22+Q23+Q24+Q25+Q26+Q27+Q28+Q29+Q30 + Q31+Q32+Q33+Q34+Q35+Q36+Q37+Q38+Q39+Q40 + Q41+Q42+Q43+Q44+Q45+Q46+Q47+Q48+Q49+Q50-1))),control=control,data=ToyDat,method='ML') summary(rf_model1) #
What if we add the PRS2, contains 1/5 of the genetic info.
So sdb estimated will be less than true value of 1 but greater than zero
# rfmodel2=lme(pheno~PRS2, random=list(unit=( pdIdent(~Q1+Q2+Q3+Q4+Q5+Q6+Q7+Q8+Q9+Q10 + Q11+Q12+Q13+Q14+Q15+Q16+Q17+Q18+Q19+Q20 + Q21+Q22+Q23+Q24+Q25+Q26+Q27+Q28+Q29+Q30 + Q31+Q32+Q33+Q34+Q35+Q36+Q37+Q38+Q39+Q40 + Q41+Q42+Q43+Q44+Q45+Q46+Q47+Q48+Q49+Q50-1))),control=control,data=ToyDat,method='ML') summary(rf_model2)
Not a question, just wanted to mention that it's Lydia Rader's birthday today and we would all sing happy birthday if we were meeting in person 🥳

*Thread Reply:* Conor - you play guitar. You really should sing happy birthday to her at the start of session B!
*Thread Reply:* I think she is on session A, so she might miss out on the whole performance
I am unclear about how to explore the files in the LD score regression tutorial. The answers dont tell you what to do at all so I dont know how to get the answer for example Q1 "What is the number of reference SNPs?" How do I find that out?
*Thread Reply:* (Please correct me if im wrong) The content of the files are the number of reference SNPs on each chromosome to calculate LD scores. So if you just follow the code "cat /data/LDSCREF/ldscores/.M" you could see 22 rows of numbers, each corresponding to the content of a single file, and just add them up. Or you could follow the code in the answers "wc -l /data/LDSCREF/plink_files/bim" which is to check the number of reference SNPs from the plink files.
*Thread Reply:* when you say you add the numbers up, which numbers? This is what I get: workshop-48:~/LDSC_practical> ls -l /data/LDSC_REF/ldscores/**.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.10.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.11.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.12.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.13.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.14.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.15.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.16.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.17.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.18.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.19.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.1.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.20.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.21.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.22.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.2.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.3.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.4.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.5.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.6.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.7.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.8.l2.M -rw-r--r-- 1 root root 7 Apr 27 2016 /data/LDSC_REF/ldscores/LDscore.9.l2.M
*Thread Reply: oh I get the numbers with your code but not with the code given in the document.... workshop-43:~/LDSC_practical> cat /data/LDSC_REF/ldscores/*.M 510501 493922 480110 366200 324698 287001 316981 269222 285156 232363 779354 221626 138712 141123 839590 706350 729645 633015 664016 589569 549971 438106
*Thread Reply:* The code given in the question session is to list the files with names ended up with '.M' under the pathway.
I'm still confused about what the "heritability" SNPs are. The references SNPs are used to get the LD scores for each of our regression SNPs, right? The Regression SNPs are the SNPs for which we have GWAS betas right? So it seems to me the only two things we need to run LDSC are those two files: We regress chi-square for the regression SNPs on their LD scores calculated using the reference SNPs. So What are the "heritability" SNPs?
*Thread Reply:* Indeed - as you say, the SNP association statistics and the LD scores are the only things required for LD score regression. So, there are not heritability SNPs rather what LD score regression is doing is estimating the heritability for the entire set of SNPs included in the calculation of the LD scores [not just the SNPs in the association analysis].
*Thread Reply:* Sorry, it's still not clear what "heritability SNPs" are. In the Day4 google document, it states, "LDSC analyses consider 3 sets of SNPs...Reference SNPs...Regression SNPs...Heritability SNPs.." What are the "Heritability SNPs"??
Is it possible to impute sumstats without individual level data? Say you're doing a cross-trait analysis on a few traits and you have GWAS sumstats for each trait. You might want to compare signal by looking at the same SNPs between traits, but the overlap of SNPs can drop quite rapidly if you are comparing several traits. Is there some way to impute SNPs to sumstats such that the traits have a larger overlap of shared SNPs?
*Thread Reply:* Yes, but it’s not the better option: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5983877/
*Thread Reply:* Are there any drawbacks with imputing summary statistics compared to just going with the 'original' overlapping SNP set? In e.g. the 'Genetic Architecture of 11 Major Psychiatric Disorders' paper, did you use this procedure there? Why/why not?
*Thread Reply:* When you “need” specific SNPs in an analysis the first choice is to go back to the raw data and impute, that’s not always feasible or worth the trouble to you or even possible, in those cases you can and should consider summary statistics imputation.
For genetic correlations, I don't really understand what the --ref-ld-chr and --w-ld-chr flags do? What is the difference between the ".ldscore.gz", ".M", ".M550" and "weights" files? Are these files usually precomputed or based on our own data? Thanks!
*Thread Reply:* @Benjamin Neale - I think this another one for you or @Steven Gazal

*Thread Reply:* Hi! • LD scores are computed on Europeans of 1000G, because (in most of the cases) you do not have sequence data from your GWAS. So yes you can use this data on all the European GWAS you are analyzing. In ldsc, you provide LD scores through the --ref-ld-chr option. • Regressions need to be performed on independent observations. As you still have LD between your SNPs in your GWAS data, you need to correct for redundant information while performing the regression. This is why you are using weights, that are proportional to 1/LD scores computed on SNPs used in the regression. In ldsc, you provide weights through the --w-ld-chr option. Hope this helps!
Hi! I am quite lost after Day 4's videos and practices...(I don't understand 90% of the content on Day 4). During the session B practice hours, I tried to hang in there and just observed everything that my roommates did without interrupting their work and discussion, but I wonder: will that be possible to put some of the beginners like me together in the same room with a teacher for session B's practice hours? Thank you!
*Thread Reply:* Hi - we're going to try something a little different on day 5 so hopefully this will help. One of the benefits of having mixed experience groups working together is that the more experienced people also learn a lot by helping people with less experience so please talk with your group as you go through the practicals. But if it's really not working for you come back to the main room and we'll work this out.
*Thread Reply:* We’re getting and reading your feedback. If there are actionable things we can do to improve the workshop experience for students, we’ll try to do so. Stay tuned
Hi! I'm very new to genetics so this might sound very amateur. I was looking a .bim file and there were some SNPs with multiple bases for either A1 or A2. For example: RSID A1 A2 rsXX TCGAA T rsXXX TA T Why is that so? Are they indels? Do we usually keep these SNPs then or do we drop them before doing further analyses? Thank you!
*Thread Reply:* Hi - Yes these are insertion/deletion polymorphisms aka indels. You can keep these variants and analyses them in the same way you would analyse a SNP
Can I include several Polygenic risk scores that are correlated, into the same (regression)model as independent variables (for example anxiety and neuroticism or educational attainment and cognitive skills)? Or will I then get a problem with over-controlling?
*Thread Reply:* you can, the question I would have is: "what are you trying to estimate?' is your goal to estimate the magnitude of the effect of one of these PRS? is your goal to estimaste their joint effect? ITs important to realise when "controlling" for a PRS you aren't controlling for the entire genetic effect but only for the part tagged in the GWAS, which depends heavily on the sample size of the GWAS.
*Thread Reply:* so control for a PRS derived from a 50.000 person GWAS isn't the same as controlling fr a PRS from 1.000.000 person GWAS. there is a pre-print describing how you could deal with measurement error in PRS: https://www.biorxiv.org/content/10.1101/165472v1
*Thread Reply:* there is also this work (its written a bit econometrically if that makes sense) which argues you should often try and use 2 PRS for a singel trait to account for measurement error: https://www.biorxiv.org/content/10.1101/2021.04.09.439157v1
*Thread Reply:* If the goal is to estimate the magnitude of the effect of one of these PRS, will it still be ok/wise to include correlated controls?
*Thread Reply:* including other PRS for correlated traits will change the meaning of the effect size you estimate though. so think trough what you want to know. Do you want to know increase in outcome given change in the PRS, or change in the outcome given a change in the PRS conditional on the other PRS? so whether its ok/wise depends on what you are after.
In the Imputation video, several imputation servers (e.g., the Michigan server) were mentioned. How are they different? Which one would you recommend more strongly? or it does not matter which server you use?
*Thread Reply:* Your choices may actually be shaped by local privacy laws and similar considerations more then by the quality of the server. That being said there may be real quality differences but I don’t have the expertise to express a strong preference.
*Thread Reply:* The quality of imputation depends mostly on the number of individuals in the reference panel and how well they match to your sample ancestrally. So try to find a ref panel that maximizes those two. In my lab, we’re finding that the TopMed sequence data outperforms the HRC by quite a lot, and especially so for non-EUR samples
*Thread Reply:* Note that TopMed seq data is publicly available through dbGaP, so you could download it and use it locally, but you can also use their imputation server which is modeled after the Michigan one: https://imputation.biodatacatalyst.nhlbi.nih.gov/#!
*Thread Reply:* I don't recommend TopMed imputation for GWAS - you gain <1M extra markers that you can use for GWAS and end up with ~300M markers you can't use for a GWAS. It's also on build38 which makes everything an order of magnitude more difficult.
*Thread Reply:* I somewhat disagree Sarah - it depends on the purpose. If you care about rare variants, we’ve found TopMed imputation to be significantly better than HRC - e.g., r2 of 70% vs. 40% for MAF of some bin (I won’t look up right now)
*Thread Reply:* for GWAS in most cohort sizes <80K you don't gain maybe 1M extra markers
Any recommendations on what genotyping arrays to use for Latinx/Mexican American samples?
*Thread Reply:* Again, a question @Benjamin Neale could answer better but the MEGA Illumina array is designed to capture maximal information across ancestries (and Latinx groups are actually an admixed group of Native American, European, and African, roughly in that order but with a lot of variation between individuals). There may be others than MEGA but that’s the one I’m familiar with
*Thread Reply:* Great question. That depends on your budget. Another greqt option could be low coverage sequencing: https://www.sciencedirect.com/science/article/abs/pii/S0002929721000963
This is probably a very naive question, but I was wondering how much does it cost to genotype (GWAS scale) one saliva sample? and for people who wants to incorporate genetic data into their research but is not a geneticist (don't do genotyping), how/where to they typically get their DNA samples genotyped? Are there recommendations for good genotyping facilities that offer genotyping services for researchers?
*Thread Reply:* I’m probably not the best person to answer this bc I don’t collect my own data or do genotyping, but the cost of array genotyping is very cheap now - say $40 neighborhood. Most of the cost will be in collecting the phenotypes and saliva. Most researchers send their data directly into Illumina or Affy I believe. @Benjamin Neale would the best person to answer this with more detail
*Thread Reply:* It depends - Matt's estimate isn't too far off, but there are economies of scale that change the cost
*Thread Reply:* in terms of where to get things genotyped - Thermofisher acquired Affy and now provide that in a fee for service way as Matt suggests - I think Illumina does as well, but I am unsure. There are genotyping facilities, often in academic settings as well
*Thread Reply:* So, Mr. Wormley and I discussed this over the weekend. For a sample of ~1000 participants, it costs approximately $100 USD/sample. This cost would be reduced as your number of participants increases. An investigator should consider additional costs: (1) collecting the DNA, (2) isolating the DNA, and (3) quantifying the DNA to the preferred specifications of the group that will do the genotyping. Many places will offer these services as add-ons (and charge a little extra). Some investigators will choose to address these preliminary steps with a lab within their organization to save on costs and ensure that they have full control of their original samples as well as storage of those samples. Some people I work with have had genotyping done with RUCDR https://www.rucdr.org/ and BGI https://www.bgi.com/us/
Hi Adrian, Thank you for a nice introduction lecture on polygenic scores. For the tutorial today I was hoping to learn and practice how to calculate PRS and was a bit disappointed that it was not part of the tutorial. Do you have scripts for calculating PRS using the various methods (e.g., PRS-CS) that you can share? Thank you very much!
*Thread Reply:* Hi - Sorry we didn't have time to go into all the code and methods today (you could spend an entire week on this). The code and steps for making a PRS with C+T are in the workbook - much of the initial code needs to be customised based on the format of the summary statistics that you have access to.
*Thread Reply:* Hi, I understand your frustration. Running a PRS from scratch can be quite time consuming so it has been challenging to implement a full practical. I am happy to share my examples and scripts with you. However, you need to understand that most of these are very specific to the sample and setup you have. For example the pieces of code I am sharing with you are based on a PBS scheduling system for a cluster. I am sharing with you snippets of SBayesR and clumping + thresholding PRS.
*Thread Reply:* Again these are examples, but the best way to learn is to go through the help of your favorite method and try implementing your own steps. I will bring up your comments to the faculty to assess implementing a simple but comprehensive PRS practical in the future.
I've heard people say "PRS has the potential to inform personalized treatment/intervention/prevention" or the like. It sounds great, but I often wonder what that really means, and I have a hard time understanding how it would actually look like in practice, particularly for complex psychiatric traits (e.g., substance use disorders, depression, etc.) where PRS now predicts only a very very small amount of variance. Even when the predictive power of PRS is significantly improved (e.g., 30% - if that ever happens), what do we really mean by "personalized" treatment/intervention? Will we screen people based on their PRS and decide whether or not they get to be included in a certain treatment/intervention? Look forward to hearing your input, Thanks!
*Thread Reply:* It depends on the use case, of course - among early demonstrations of potential utility are perhaps most prominent in cardiovascular disease (e.g., https://pubmed.ncbi.nlm.nih.gov/30309464/) and there are arguments about the potential in cancer screening, particular breast cancer as age isn't terribly predictive for breast cancer risk
*Thread Reply:* the traits you mention are indeed some of the hardest to envision a PRS being useful for. some work by Naomi Wray very clearly outlines the realistic limitations in psychiatric applications: https://pubmed.ncbi.nlm.nih.gov/32997097/ and https://pubmed.ncbi.nlm.nih.gov/33052393/ Psychiatry isnt going to be the first domain in which PRS are applied, PRS for breast cancer, combined with rare variant screening or PRS for cardiovasscular disease combined with classical risk factors seen more reasonable candidates for clinical application in the coming decade.
*Thread Reply:* This is a good question. We've essentially posed it as the RCR question for Day 7: What should PRSs be used for? Clinical risk prediction? Lifestyle choices; preventative medical intervention; insurance assessment; how about educational attainment prediction and assignment to school programs? Or prenatal testing and embryo selection? Or partner choice?
I wasn't able to stay for the whole practical on day 4 when we talked about intercepts, so this may have been discussed more then, but I have never really been sure on how to interpret intercepts in ldsc. Does anyone have an explanation of this or a resource? I have also read that an intercept of >0 when you correlate two datasets indicates sample overlap. Why is this the case?
*Thread Reply:* Hi- The formula for LD score regression (univariate) is given in equation (1) in the Bulik Sullivan et al. (2015) (LD Score regression distinguishes confounding from polygenicity in genome-wide association studies (nature.com) paper. It shows that the expected value of the chi-square statistic given the LD score for a variant of interest E(chisq | lj) = (Nh^2/M)lj + Na + 1 Where chisq is the chisquare test statistic, N is the sample size, h^2 is the SNP based trait heritability, lj is the LD score for variant j (i.e. a measure of how many other variants SNP j tags), M is the number of markers and “a” a measure of bias contributed from e.g. population stratification, crytpic relatedness etc. You can basically think of this equation as a linear regression where we are regressing chisquare statistic on ld score across all the markers typed across the genome. The first term in the equation above is the slope of the regression (Nh^2/M), and the next two terms are the intercept (i.e. Na + 1). In other words, when there is no bias from cryptic relatedness/population stratification etc the “a” term will equal zero and so the intercept will equal 1. Therefore, inflation of the intercept above one suggests the presence of one or more sources of bias (cryptic relatedness/population stratification). The formula for bivariate LD score regression (where we estimate the genetic correlation across traits) is given in equation one in An atlas of genetic correlations across human diseases and traits (nature.com). It is: E[z1j, z2j] = (sqrt(N1N2)rhog/M)lj + rhoNs/sqrt(N1N2) Where N1, N2 the sample size of N1 and N2, rho-g the genetic correlation, lj the LD score as above, rho the phenotypic correlation between the traits in the Ns overlapping samples, M the number of markers, Ns the sample overlap. Again looking at the form of the regression, you can see that the intercept is rhoN_s/sqrt(N1N2) and so a function of sample overlap. Dave
Hi Michel and Andrew, thank you for the really clear videos and lectures on genomicsem. If I were interested in looking at the shared and unshared genetics effects for two traits and the SNPs underlying these effects (and neither of these traits is causal for the other) could a multivariate GWAS be used with a common factor model for these two traits and then split SNPs into 'shared' and 'unshared' genetic effects based on their q-statistic?
*Thread Reply:* that seems like a reasonable approach to me! I'll just note that with a common factor for two traits, you'll need to specify that the factor loadings are equal across the two as the model will not be identified otherwise (i..e, you'll be using more degrees of freedom then are available to you). to set the factor loadings to equal you would include an equality constraint by labeling them with the same parameter (e.g., F1=~aX1+aX2).
*Thread Reply:* So I (we and likely others) have spend a lot of time thinking of this problem. If you have no idea of the causal relation (or think you know neither trait causes the other) then you best thread carefully right? So I would also do that here, and ask you a counter question: what is your goal with the set of SNPs you want to select? if the goal is in the realm of PRS/rg then i'd give other recommendations then if the goal is some kind of locus specific functional follow up. What I can say for certain is that splitting up loci this way is non trivial as you'll perform multiple inferences, first you test whether Q is significant or not, then you presumably test whether any of the effect (on the factor or on trait A or B) is significant. your hypothesis tests are dependent, als how are you integrating this info over SNPs? neighbouring SNPs might end up in different "categories" but be in LD? I see the value in what you want to do, but I think its a hard problem, it these are case/control traits also consider case/case GWAS to supplement your analysis: Peyrot Alkes P…
*Thread Reply:* Thank you both for your responses! I would interested in trying to split the SNPs into shared and unshared effects and then doing functional follow ups / gene-set analyses on these two groups of SNPs. For this kind of goal what might be the best/most advisable approach?
how long will we be able to view the information on here (Slack), will it remain indefinitely?
*Thread Reply:* We haven't decided the exact amount of time it will stay active, but I will make an archive of all of the public channels available on a website.
In other words, you will be able to continue to ask questions here for a week+ or so, but will able to refer to the answers and discussions indefinitely.
What are the implications of running a GWAS by subtraction where neither trait is causal of the other (e.g., SCZ and BIP), and what problems arise when doing so? If it is not a valid approach, are there other ways you would suggest of approaching this type of research question?
*Thread Reply:* Good Q, ill try and circle back to it after the practical!

*Thread Reply:* Any follow-up on this Q at today's practical? I must have missed it.
*Thread Reply:* So @Benjamin Neale sort of commented on this, the GWAS by subtraction is only a model, based on the average correlation across the genome, and we should expect for any pair of traits that some SNPs act on both, some only on one of the traits and some in opposite directions on the two traits. if the relation between a pair of traits is causal, or possibly causal then the effects on the cause add to the GWAS of the outcome and subtraction makes sense, it their is a pair of trait with a set of common (latent) causes as well as unique influences then a different model makes sense. a some you could fit is a common factor and Q and then inspect the Q hits and figure out their effect (only on one or on both but discordant direction). You could also do a GenomicSEM GWAS with an effect on the common factor, a GenomicSEM GWAS with an effect on trait A and a GenomicSEM GWS with an effect on trait B, and model average the results (see for model averaging:https://research.vu.nl/ws/portalfiles/portal/123186830/Multivariate_genomewide_analyses_of_the_wellbeing_spectrum.pdf) or try alternatives like LAVA: https://www.biorxiv.org/content/10.1101/2020.12.31.424652v2. A paper I love, which does reflect the philosophy that really SNPs can influence a pair of traits in many ways is LHC-MR (see: https://www.medrxiv.org/content/10.1101/2020.01.27.20018929v2) I recommend it to anyone that wants to expand their horizon when it comes to multivariate analysis. Critical to realise is that once you have hits of interest its not really a huge statistical problem to go and figure out whether they act on one trait both traits or in opposing directions.
I am potentially interested in using a genomic p-factor in a study to look at genetic correlations of the p-factor and other phenotypes. Are there genomic p-factors available for download similarly to how we can download summary stats for individual psychiatric disorders, or is it necessary to derive my own genomic p-factor through genomicSEM? I know many different traits can go into a p-factor, but I was unsure if there is a particular one that is being used more commonly.
*Thread Reply:* we make the summary statistics for the five indicator (major depression, anxiety, schizophrenia, bipolar disorder, and PTSD) p-factor from the original Genomic SEM publication available in this box link: https://utexas.app.box.com/s/vkd36n197m8klbaio3yzoxsee6sxo11v
*Thread Reply:* I would note that as we've expanded our psychiatric models to include more disorders, we've found less evidence for a p-factor at different levels of analysis. For example, we find that most SNPs do not fit a p-factor model when we include 11 disorders. you can read more about those results here if interested: https://www.medrxiv.org/content/10.1101/2020.09.22.20196089v1
What is the recommended method for running a GWAS in samples with known relatedness?
*Thread Reply:* Good question - it depends on the nature of the relatedness and the phenotype. For continuous, particularly normal distributed traits, mixed models, with the inclusion of the genetic relatedness matrix to model the relatedness work very well [see the bolt-lmm (https://alkesgroup.broadinstitute.org/BOLT-LMM/BOLT-LMM_manual.html), or warpedlmm (https://www.nature.com/articles/ncomms5890) manage that kind of analysis well. For binary outcomes, I would say the landscape is less clear [there are complexities around how exactly these models work] - that being said, the recent regenie paper [https://www.nature.com/articles/s41588-021-00870-7] - is another way to perform GWAS that may be effective, depending on the context.
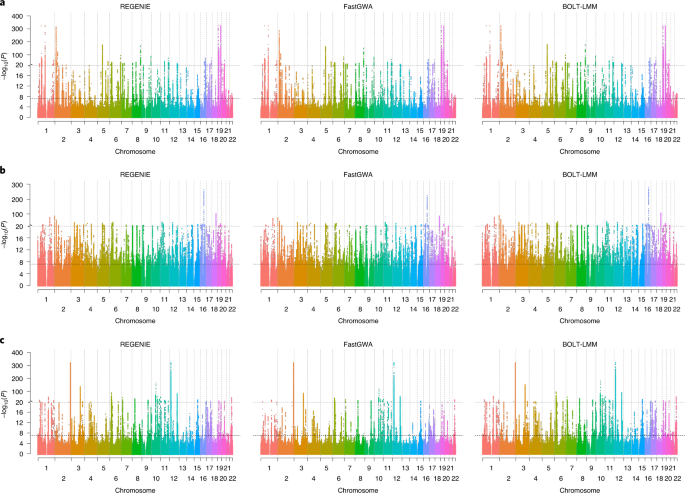
*Thread Reply:* FWIW, I am aware of two papers concerning LMMs for dichotomous traits: https://www.cell.com/ajhg/fulltext/S0002-9297(15)00105-6 and https://www.nature.com/articles/nmeth.3285 .
Is there a recommended minimum number of non-European individuals clustering together based on the first genome-wide PCs for conducting an independent non-European GWAS?
*Thread Reply:* all the factors that influence the performance of GWAS hold so it depends on the trait [continuous traits perform better than binary or discrete traits] and the frequency threshold [more people enable going rarer] - in the UKB analyses across the different ancestry groups, you can see how there are variable numbers of traits for the different subgroups https://pan-dev.ukbb.broadinstitute.org/docs/technical-overview
Will it be possible to ask questions in the weeks following the workshop? I feel there is a lot to unpack and I might return to the material shortly after
*Thread Reply:* Sorry for the delay in responses, I was hoping to put in something more concrete.
All public channels on slack will be archived on the web in a browseable location. This will allow you to look at existing content.
Current students will be converted to guests and they will only have access to a single public channel. (The workshop pays for full members, so we can't keep all of the students as full members indefinitely, but single channel guests are free.)
So, following the workshop there will be a single channel where you can ask questions.
However, the faculty and tutors aren't being committed to follow that channel for any specific length of time. Hopefully, many will monitor slack, as they use it day-to-day anyway. It is the recommended first step in asking questions.
Can the models presented in day 7 (mendelian randomisation) also be fitted within the SEM framework?
*Thread Reply:* some of them yes, or as a Bayesian regression: https://rpsychologist.com/adherence-analysis-IV-brms
*Thread Reply:* doing so can add complication but also add lots of flexibility
*Thread Reply:* Great question! Yes! 🙂 Some of the assumptions underlying SEM via Maximum likelihood and MR via e.g. two stage least squares can be a little different so important to bear this in mind when fitting models.
*Thread Reply:* Yes - and of course most SEM packages also have least squares fitting functions, whose assumptions are less restrictive than MVN.
*Thread Reply:* It always seemed to me that SEM using lasso, ridge and elasticnet estimators would be very useful in the context of single sample MR. basically estimate the pleiotropic paths (all of them) but impose a penalty on them.
I have been fielding a few questions around ML/AI in genetics and how to think about good use of AI/ML and bad use of AI/ML. So for good use - phenotype construction from imaging is a great example [here's an example of https://www.biorxiv.org/content/10.1101/2021.02.05.429046v1.full that uses deep learning to extract traits from cardiac MRI] - further ML methods have been used for predicting regulatory effects of variants [https://www.nature.com/articles/s41467-020-18515-4 is a good example] although the extent to which these predictions are informing on exactly what the variant is doing is unclear - the last line of Dey et al is
We conclude that deep learning models have yet to achieve their full potential to provide considerable unique information for complex disease, and that their conditional informativeness for disease cannot be inferred from their accuracy in predicting regulatory annotations.
For bad use of ML/AI - we mentioned Skafidas et al. [https://pubmed.ncbi.nlm.nih.gov/22965006/] which builds a predictor for ASD based on GWAS and is likely picking up some amount of confounding bias [we wrote a response - https://www.nature.com/articles/mp2013125] and this example illustrates that ML/AI applied can easily be led astray by confounding biases [e.g., population stratification]
So as with anything, it depends and it helps to think about the problem you are trying to tackle.
*Thread Reply:* With ML it is essential to benchmark performance against simpler techniques (e.g., if you are performing lasso, compare it to OLS; if you are doing deep learning, compare it to lasso).
Deep learning has a lot of promise theoretically, as a neural network with enough layers can approximate any function (the Universal Approximation Theorem). A big challenge in deep learning for genetics is finding a good way to represent the data as it is fed into the neural network (this is called an "embedding"). There are a lot of bad models out there were genetic data gets reformatted as an image and fed into a pre-existing deep learning model used for image recognition. This works, as the model can learn from the inputs almost no matter how it is structured, but it is not optimal (as the 2D structure of the genetic data is meaningless, unlike with an image, where neighbouring pixels probable represent the same object)
When will we receive the username and password for today's practical in Hail?
*Thread Reply:* At the beginning of the session! Giving you this info now won’t let you log in ahead of time, since the workshop system isn’t turned on yet.
Just to let the tutors know, there a lot of parts of today's videos where the video skips entire sections
*Thread Reply:* Thank you for this note! If you could let us know which sections, we can correct this!
What is the difference between eQTLs and variants that influence regulatory function. or... what is the difference between regulation and gene expression (Day 10 topic)
*Thread Reply:* So an eQTL stands for expression quantitative trait locus - I would say it’s current usage is heavily loaded toward baseline expression - tho some efforts to evaluate the eQTLs in dynamic cell states (https://elifesciences.org/articles/67077) have started. Additionally sometimes splice QTLs (sQTLs) are interrogated (more asking the question of which isoform of the gene is transcribed and whether genetic variation influences that) - GTEx has some resources on that - which is a different kind of regulatory process. There may also be variation that influences trafficking of the gene product which you could conceptualize as a regulatory process or even post translational modification of proteins that genetic variation might influence. So I think, loosely, regulatory variation is sometimes used as shorthand for anything that it’s not obviously coding in consequence but sometimes is referring to gene regulation more explicitly but as the comment illustrates, what is in gene regulation is less clearly defined.
We were advised not to use genomic control in a GWAS if GWAS sum stats are to be used for LD score regression. PLease would you mind elaborating on what is meant by using genomic control, thanks! (Day 3/ day 5)
*Thread Reply:* So Genomic control is a technique used to “correct” GWAS summary data for perceived inflation due to population stratification. It relied on the average test statistic across all SNPs to correct the inflation in those test statistics. the logic is that most inflation would not be due to true genetic effects. It has fallen in disfavour because 1. for polygenic traits most inflation in test statistics is actually due to true heritable signal (if you do your GWAS/QC properly!) and 2. the LD score regression intercept is a more direct (yet still imperfect!) measure of inflation due to population stratification then the statistics hitsorically used for genomic control. if you first apply genomic control then LD score regression results will be biased because you alter the test statistics in your GWAS. genomic control has a wiki page actually: https://en.wikipedia.org/wiki/Genomic_control (not vouching for the contents…)
*Thread Reply:* Basically, genomic control is an outdated way to control for population stratification, relatedness, and other confounders. It was predicated on the idea that traits would be influenced by a small number (e.g., 100s) of SNPs and that if you saw 10's of thousands of SNPs having inflated association, that association must be due to uncontrolled confounders like population stratification. Now that we have better ways to control for these (esp. mixed effect models, but also ancestry PCs), it has become obsolete.
Are there specific guidelines for including the X chromosome in a GWAS and meta-GWAS? Is it recommended and what are the main aspects to consider?
*Thread Reply:* The inclusion of X chr in GWAS is tricky. To my mind, the main question in the X chr analyses is how to code for X chr (so we take into account the allele dosage effect between males and females). One option could be to run the analyses in PLINK and code the male X chr genotypes as 0/2 instead of 0/1. In PLINK, this would be the following command (for binary trait, change the regression to linear if needed):
plink --bfile FILE --logistic --ci 0.95 --xchr-model 2 --covar your.covar.file --covar-number your.number.if.needed --out OUT_FILE
(more info on this is here: https://www.cog-genomics.org/plink/1.9/assoc#linear, see —xchr-model flag).
In covar file include sex and PCs (and anything else your data may need).
*Thread Reply:* For a META-ANALYSIS inclusion of X is the same as autosomes as long as all groups have used the same type of model for the X analysis you can use and inverse variance weighted analysis. If there is any chance that the X analysis differed between groups the beta and se will be on different scales and you should use N weighted (Zscore) meta-analysis
Are there any databases/lists that states the psychological/personality/behavioral traits for which there exist PRS’s of good quality?

*Thread Reply:* Not as many behavioral traits here, but the PGS Catalog is a great resource for published polygenic scores http://www.pgscatalog.org/browse/traits/
When including PRSs in regression models, will the strength of the predictability/heritability of the specific PRSs influence the size of the estimate/whether there’s any association relative to other controls, or is this accounted for in the model automatically? And, if including PRSs as controls for an association between two phenotypes, will the impact be small because the “effect” of only one PRS alone will never give a large “effect”?
*Thread Reply:* If you use multiple regression with all predictors entered in 1 step the model fits all effects simultaneously and so betas are relative to all predictors. (Not using the word control in that sentence as using this term can be confusing when the regression involves genetic analysis).
*Thread Reply:* re using a PRS to "control" for a non measured variable - I think the answer is it depends. Many PRS can predict ~5% percent of the variance this is often the effect size you see for variables such as Age and Sex which are commonly included in many analyses
Is there a recommended automatic procedure for clustering individuals by ancestry and please could you provide reference for a pipeline? Standard approaches like using k-means, random forest, standard deviation from reference panel’s centroids...
*Thread Reply:* Great question - the gnomad team has some pretty extensive documentation on how it approaches ancestry assignment: https://gnomad.broadinstitute.org/help/ancestry - there a random forest approach is used. This follows from Rfmix https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3738819/ that we use in tractor (https://www.biorxiv.org/content/10.1101/2020.05.17.100727v1) to get at local ancestry estimation
*Thread Reply:* this github could be useful (it is from gnomAD):
*Thread Reply:* https://github.com/broadinstitute/gnomad_methods/blob/master/gnomad/sample_qc/ancestry.py
In today's lecture 2 introducing SAIGE, it refers to "centered and scaled" genotypes. What does that mean? • What does genotype data look like before being centered and scaled, 0/1/2 (effect allele counts per individual at each locus)? • Does center and scale just mean "subtract mean and divide by standard error" using these 0/1/2 genotypes for each locus? • Does it matter for this procedure that different loci have different minor allele frequencies? • Does it matter for this procedure if some loci are not in Hardy Weinberg equilibrium?
*Thread Reply:* it means that genotypes are converted to z-scores with mean=0 and var=1. The distribution of genotype scores will still look the same (binomial) but the mean & var will be different than before (i.e., will be identical across all SNPs). It doesn’t matter that some SNPs have different MAF or are not in HWE (so long as the empirical and not the expected SD is used in the denominator of the z)
*Thread Reply:* Thanks! So like this? > locusA <- c(0,0,0,0,0,1,1,1,1,2) > locusB <- c(0,0,0,1,1,1,1,2,2,2) > scale(locusA,center=T)[,1] [1] -0.8581163 -0.8581163 -0.8581163 -0.8581163 -0.8581163 0.5720776 0.5720776 0.5720776 [9] 0.5720776 2.0022714 > scale(locusB,center=T)[,1] [1] -1.224745 -1.224745 -1.224745 0.000000 0.000000 0.000000 0.000000 1.224745 1.224745 [10] 1.224745
Is there any way that we (as a student) can keep in touch with other students after the workshop is ended? Like, if people want to share their name and contact information with others, maybe we can come out with a list to share with everyone? :)
*Thread Reply:* Next week I'll be sending out information about how to download your files, and other things. I'll include information about an email list with that.
Short term, slack will be available for a few weeks. Longer term, students will have access to a single guest channel on slack, where they will be able to continue to interact.
*Thread Reply:* For those on facebook there is also a workshop group and this can be good place to keep connection https://www.facebook.com/groups/twin.workshop
I have a question about the gene manhattan plots (day 10, video 2). @danielle.posthuma I have not seen many gene manhattan plots in publications. Do you think there's a reason for this? Thanks!
*Thread Reply:* I think your observation is correct- i don’t think there is a real reason behind this though. We sometimes add them in the supplement. Perhaps people find it confusing to display two similar plots, one that contains SNP p-values and the other gene P values, so in practice we often display the SNP Manh plot and a gene-based Table when the number of Figures is limited in a journal for example. Other than that - no valid reason i guess!
Thanks for the informative lectures on gene-set analysis. I was wondering if it is necessary to control for genetic background? Or is this what the competitive analysis does?
*Thread Reply:* Yes, if you mean background (SNP-) heritability than that is indeed what the competitive analysis does
Q re Day 10: with functional mapping and everything involved in day 10, will we need ancestry specific datasets/ approaches . we know that MAF differ across ancestries. Do things like Chromatin structure and gene sets differ by ancestries @danielle.posthuma
*Thread Reply:* I would not expect chromatin structure to differ between populations, but eQTL associations are expected to differ as a function of LD differences between populations
*Thread Reply:* So it would indeed be highly valuable to have eQTL information across different populations
will there be a certificate of completion for the workshop? And is there a document with an outline/summary of all the topics covered?
*Thread Reply:* Yes, certificates will be sent out (today I believe). The topics won’t be listed on the certificates themselves but they are of course listed on the workshop website
Hello, question about working with files in the bash shell/Linux terminal (something I’m rather new to). I have a dataset where rsIDs aren’t available, so I am deriving them based on available chromosome and map positions. Code-wise, how would I go about concatenating my columns, e.g. so I now have a column with CHR:SNP:A1:A2, made by combining some of the other columns and separating the values with a colon? I tried loading the bim file into R to do it there, but R kept crashing (I think the file is too large for it to handle, perhaps? 2.5GB) Thank you for any help you can provide! If you know of resources (e.g. websites) that go through file manipulation like this in the terminal (as well as other common commands for data merging, concatenating, adding/removing columns/rows, etc.), that is wonderful as well.
*Thread Reply:* Hi! awk is a good tool to manage data in this case. There are a few tutorials on internet that you can check. For this example, you could do something like: awk '{print $1":"$2":"$3":"$4,$0}' yourfile > newfile where $1, $2, $3, $4 would be your columns for CHR, SNP, A1, and A2 and $0 would print the rest of the columns on the file. ":" would put the colon in between.
*Thread Reply:* Probably the two primary commands to use would be either paste or join. paste is going to be faster and use less memory if the files are already row aligned. So you would do something like
paste -d: <file1> <file2> > outfile
If the files aren't matched by row, but just by some ID, like position, then join would be what to use
join -1 3 -2 2 <file1> <file2> > outfile
if position is in column 3 of file1, and column 2 of file2. join can require sorting, and uses space as a delimiter, so it can be trickier to use, and may require pre- or post-processing with something like sort , tr, or sed
Or am I not understanding, and you have one file with : delimited columns? then do like Lucía says.
*Thread Reply:* Thank you so much to you both! This is very helpful, I truly appreciate such quick responses.